
Vol.:(0123456789)
Clinical Drug Investigation
https://doi.org/10.1007/s40261-020-00981-9
ORIGINAL RESEARCH ARTICLE
First‑in‑Man Safety, Tolerability, andPharmacokinetics ofaNovel
andHighly Selective Inhibitor ofMatrix Metalloproteinase‑12, FP‑025:
Results fromTwo Randomized Studies inHealthy Subjects
KhalidAbd‑Elaziz
1
· ChristineVoors‑Pette
1
· Kang‑LingWang
2,3
· SandyPan
4
· YishengLee
5
· JohnMao
5
·
YuhuaLi
5
· BenjaminChien
5
· DavidLau
5
· ZuzanaDiamant
1,6,7,8
Accepted: 21 October 2020
© Springer Nature Switzerland AG 2020
Abstract
Background and Objectives Matrix metalloproteinases (MMPs) are proteases with different biological and pathological
activities, and many have been linked to several diseases. Targeting individual MMPs may offer a safer therapeutic potential
for several diseases. We assessed the safety, tolerability, and pharmacokinetics of FP-025, a novel, highly selective oral matrix
metalloproteinase-12 inhibitor, in healthy subjects.
Methods Two randomized, double-blind, placebo-controlled studies were conducted. Study I was a first-in-man study,
evaluating eight single ascending doses (SADs) (50–800 mg) in two formulations: i.e., neat FP-025 in capsule (API-in-
Capsule) and in an amorphous solid dispersion (ASD-in-Capsule) formulation. In Study II, three multiple ascending doses
(MADs) (100, 200, and 400 mg, twice daily) of FP-025 (ASD-in-Capsule) were administered for 8days, including a food-
effect evaluation.
Results Ninety-six subjects were dosed. Both formulations were well tolerated with one adverse event (AE) reported in the
800 mg API-in-Capsule SAD group and seven AEs throughout the MAD groups. The exposure to FP-025 was low with the
API-in-Capsule formulation; it increased dose-dependently with the ASD-in-Capsule formulation, with which exposure to
FP-025 increased in a greater-than-dose-proportional manner at lower doses (≤100mg) but less proportionally at higher
doses. The elimination half-life (t
1/2
) was between 6 (Study I) and 8h (Study II). Accumulation of FP-025 was approxi-
mately 1.7-fold in the MAD study. Food intake delayed the rate of absorption, but without effect in theextent of absorption
or bioavailability.
Conclusion FP-025 was well tolerated and showed a favorable pharmacokinetic profile following ASD-in-Capsule dosing.
Efficacy studies in target patient populations, including asthma, chronic obstructive pulmonary disease (COPD), and pul-
monary fibrosis, are warranted.
Trial registration number: www.clini caltr ials.gov: NCT02238834 (Study I); NCT03304964 (Study II).
Trial registration date: Study I was registered on 12 September 2014 while study II was registered on 9 October 2017.
* Khalid Abd-Elaziz
Extended author information available on the last page of the article
1 Introduction
Matrix metalloproteinases (MMPs) comprise a family of
over 20 zinc-dependent proteases and exhibit a wide range
of biological activities [1–4]. As a class, MMPs show struc-
tural similarities and share an array of common substrates;
however, individual MMPs have distinct functions depend-
ing on their environment [1, 5]. Delivered as inactive zymo-
gens, the pro-MMPs undergo proteolytic and non-proteolytic
cleavage activation into the respective MMPs [6, 7]. MMPs
play a pivotal role in the degradation and removal of the
extracellular matrix, which is vital for several physiologi-
cal processes, including morphogenesis, tissue repair, and
remodeling [2, 8]. More recent data from both animal and
human studies show that MMPs are also involved in various
other biological processes [9, 10], including cell migration
[11], cell proliferation [12], wound healing [13], immunity,
and inflammation [14, 15].
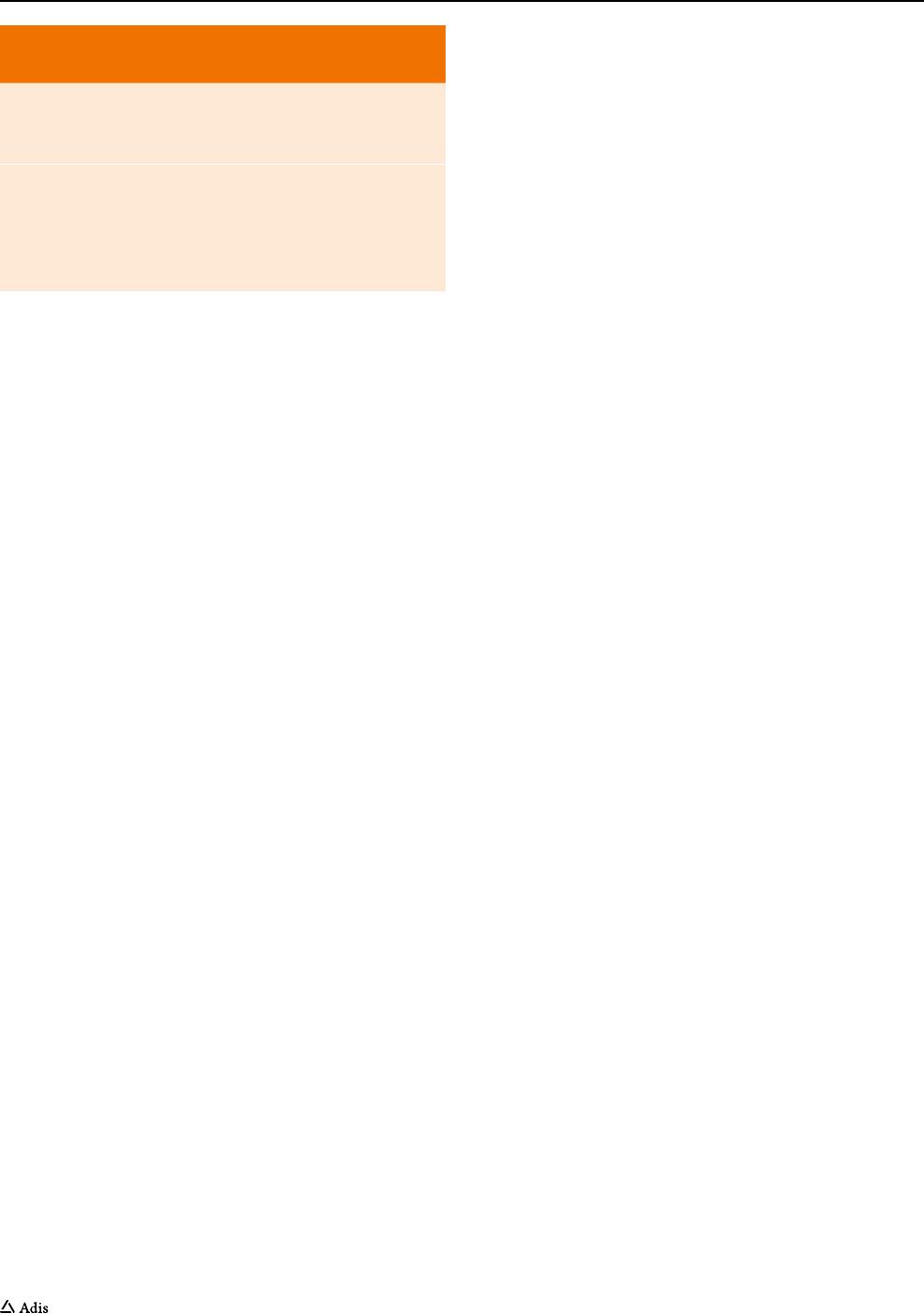
K.Abd-Elaziz et al.
Key Points
Non-selective inhibition of matrix metalloproteinases
(MMPs) has been associated with serious side effects
resulting in discontinuation of development programs.
FP-025, an oral MMP-12 inhibitor, was safe, well toler-
ated and showed a favorable pharmacokinetic profile in
healthy subjects at all single- and multiple-ascending
doses tested in ASD-in-Capsule formulation, warranting
exploration of its activity in target populations.
MMP-12 (half maximal inhibitory concentration (IC
50
):
0.04 µM), with 90-fold selectivity over the next closest
family member (MMP-2) and two to three orders of magni-
tude selectivity over the seven other MMP family members
(MMP-1, MMP-3, MMP-7, MMP-8, MMP-9, MMP-13, and
MMP-14) that were tested (unpublished data).
Currently, FP-025 is under development for the treatment
of chronic respiratory diseases. In unpublished experimental
results, FP-025 demonstrated anti-inflammatory activities
and reduced levels of tumor necrosis factor-α (TNF-α) and
interleukin-5 (IL-5) in lungs, resulting in improved lung
function and histology in a mouse model of allergic asthma.
In off-target receptor panel screen, FP-025 did not show
any important secondary pharmacodynamic interactions
or safety related pharmacodynamic issues. Safety pharma-
cology studies of FP-025 revealed no significant effects in
animal models.
Here we present the first-in-man safety and pharma-
cokinetics data of FP-025 derived as primary objectives
from two studies in healthy subjects. Secondary objectives
included evaluation of pharmacokinetics of single and mul-
tiple ascending FP-025-doses. An exploratory objective was
food effect on the pharmacokinetics of FP-025.
2 Subjects andMethods
2.1 Study Designs
Two phase I studies were performed. The first study (Study
I) was a double-blind, randomized, placebo-controlled, sin-
gle ascending-dose (SAD) study performed at Taipei Veter-
ans General Hospital, Taipei, Taiwan (Fig.2). Participants
were confined to a phase I unit from day −1 till day 2. The
second study (Study II) was a double-blind, randomized,
placebo-controlled multiple ascending-dose (MAD) study,
which also included an open-label food-effect part, con-
ducted at QPS-Netherlands, Groningen, the Netherlands
(Fig.3). Subjects were admitted to the phase I unit on day
−1 and discharged on day 10. Both studies were sponsored
by Foresee Pharmaceuticals Co., Ltd., Taipei, Taiwan and
were conducted in compliance with the current version of
the Declaration of Helsinki for medical research in human
subjects, the International Council for Harmonisation guide-
lines for Good Clinical Practice including locally applicable
regulations. All subjects provided written informed consent
prior to study enrollment.
Both study protocols, including amendments and asso-
ciated documents, were approved by the local institutional
review boards (Study I was approved by institutional
review board, Taipei Veterans General Hospital, Taipei,
Tawian; Study II was approved by Independent Ethics
Committee, Stichting BeBo, Assen, the Netherlands). Both
As a consequence of their pro-inflammatory and tissue-
remodelling activities, several MMPs are involved in the
pathophysiology of chronic inflammatory airway diseases
[16, 17], and hence, could serve as therapeutic targets. How-
ever, previous studies have shown that non-specific target-
ing of MMPs resulted in broad-spectrum inhibition of pro-
teolytic activities, inducing a variety of (serious) adverse
events (AEs) and off-target effects [18], and, consequently,
the clinical development of non-selective MMP-inhibitors
was preliminarily discontinued [19–25]. Selective targeting
may therefore offer a safer option.
Among the MMP family, MMP-12 is a 54-kDa proen-
zyme, which is processed into a 45-kDa and subsequently
into a 22-kDa active form [26, 27]. Known as macrophage
elastase, MMP-12 is predominantly detected in alveolar
macrophages [27]. In addition, MMP-12 is produced by
bronchial epithelial cells [28] and airway smooth muscle
cells [29]. In both animals and humans, this protease is
involved in type 2 inflammation as well as in tissue remod-
eling through its ability to turn over elastin during the whole
life cycle [30–33]. Accumulating evidence has suggested the
involvement of MMP-12 in the pathophysiology of chronic
inflammatory airway diseases [16, 31]. In patients with
asthma, chronic obstructive pulmonary disease (COPD),
and/or pulmonary fibrosis, the extent of airway remodeling
and/or disease severity correlates with gene expression,
local airway concentrations, and/or activitities of MMP-12
[33–37]. Recent evidence from a transgenic mouse model of
type 2 inflammation showed that eosinophil-derived inter-
leukin (IL)4/13 promoted MMP-12 release from alveolar
macrophages and was associated with alveolar destruction
[38]. Similarly, in patients with chronic inflammatory airway
disease, sputum eosinophilia was associated with increased
MMP-12 levels, which negatively correlated with lung func-
tion [38]. These data underscore the association between
type 2 eosinophilic inflammation and MMP-12 and its
potentially important role in the pathophysiology of airway
remodeling (Fig.1).
FP-025 (C
21
H
18
N
2
O
4
S) is a novel oral, non-hydroxy-
maic small molecule. FP-025 is a high-affinity inhibitor of
Safety, Tolerability, and Pharmacokinetics of FP-025
studies were registered in ClinicalTrial.gov (study identifiers
NCT02238834 and NCT03304964, respectively).
2.2 Study Subjects
Eligible participants were healthy male subjects (Study I)
or male and female subjects of non-child-bearing poten-
tial (Study II), aged between 18 and 65 years, and were
in overall good physical and mental health condition as
determined by medical history and physical, vital signs,
laboratory and electrocardiographic (ECG) examinations.
Other inclusion criteria consisted of a body mass index
between 18 and 30 kg/m
2
, a resting pulse rate between 50
and 100 beats per minute and resting blood pressure with
a systolic blood pressure of ≤ 140 mmHg and a diastolic
blood pressure of ≤ 90 mmHg. Male subjects had to use
adequate contraception during and until 3 months after
completion of the study. Main exclusion criteria consisted
of: A history of alcohol or drug abuse, or current smoker
or using other nicotine products. Use of any prescription
or non-prescription medications, herbal remedies, vita-
mins, or minerals within 2 weeks prior to the first dose (or
within five half-lives prior to inclusion for any medica-
tions ingested, whichever was longer). A positive test for
hepatitis virus B or C, or human immunodeficiency virus,
or a QT interval (according to ECG examinations after
Bazett’s correction) of more than 450 ms.
2.3 Study Medication andRegimens
Based on the No Observed Adverse Effect Levels (NOAELs)
for the 28-day toxicity studies, the human equivalent dose
for the rat yields a more conservative value of 161 mg/kg/
day. To be conservative, a safety margin of 50-fold is chosen
for the starting dose of the first-in-human study, resulting in
a starting dose of 200 mg in humans (161 (mg/kg)/50=3.22
(mg/kg), which is approximately 200 mg using a body
weight of 70 kg). Two different oral formulations of FP-025
were tested. The initial formulation was a neat active (spray-
dried API in capsule) pharmaceutical ingredient (API) in
oral capsules of FP-025 (API-in-Capsule), which was tested
at the dose levels of 200 mg, 400 mg, and 800 mg. Due to
the persistence of low exposure to API-in-Capsule formula-
tion in the first three cohorts, another formulation was used.
Subsequently, an improved oral formulation was tested,
the amorphous solid dispersion (ASD) of FP-025 (ASD-in-
Capsule) at dose levels of 50 mg, 100 mg, 200 mg, 300 mg,
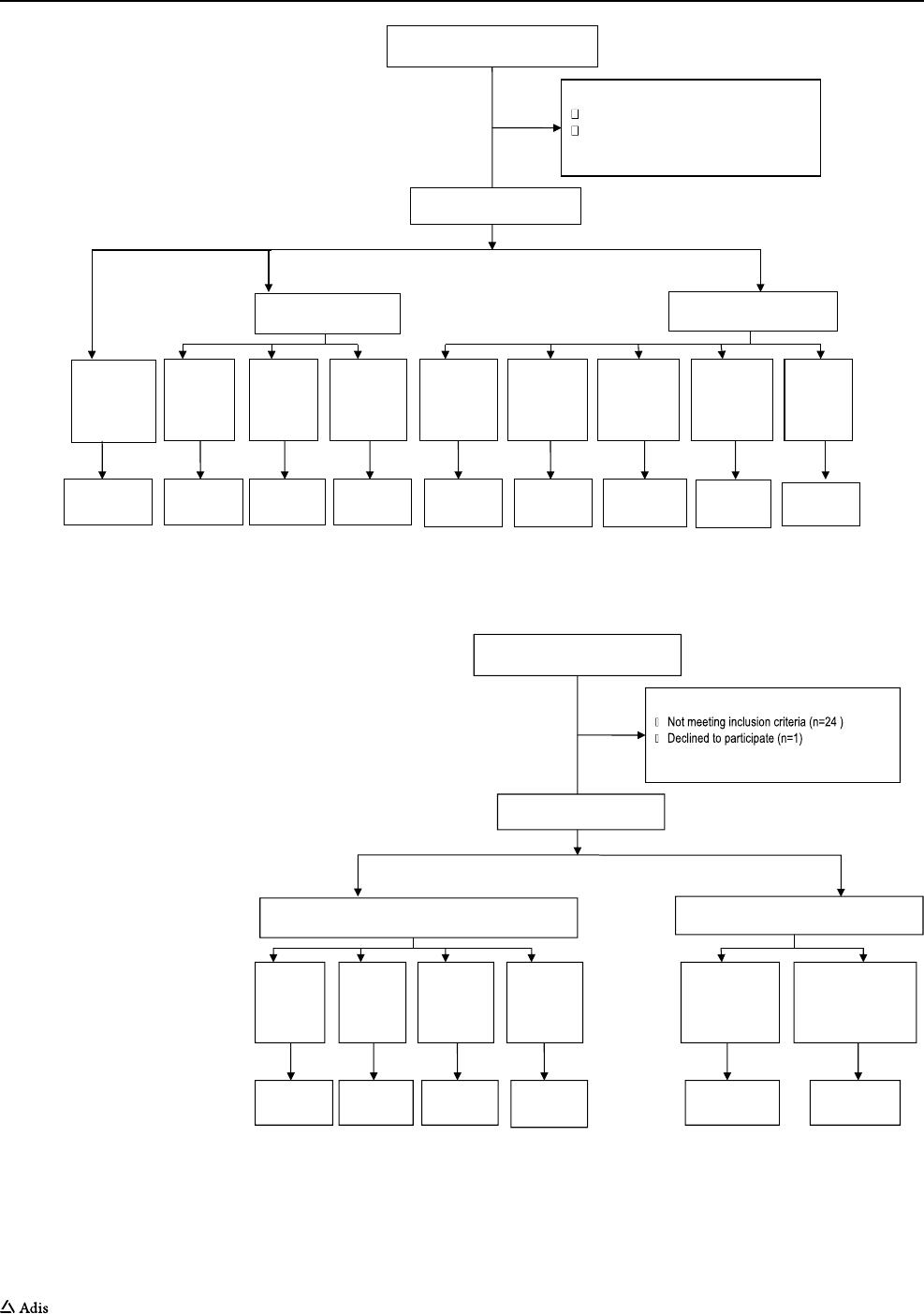
and 450 mg (Fig.2).
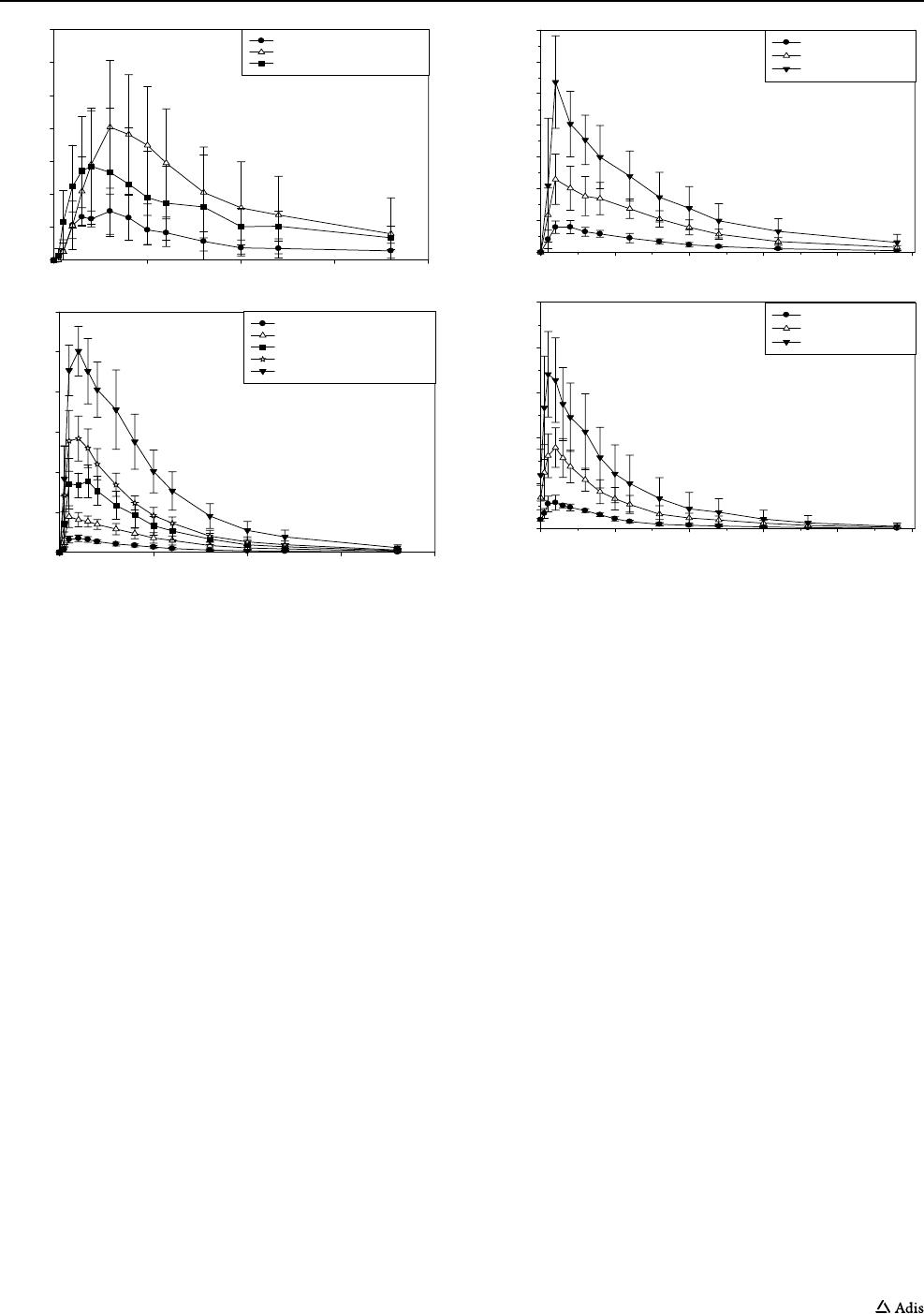
Fig. 1 Role of MMP-12 as a potential mediator of both inflammatory processes and structural changes that occur in asthma and chronic obstruc-
tive pulmonary disease (COPD). TGF transforming growth factor, TNF tumor necrosis factor, MMP matrix metalloproteinase
K.Abd-Elaziz et al.
In Study II, the following treatments were administered:
100 mg (treatment A), 200 mg (treatment B), and 400 mg
(treatment C) FP-025 ASD-in-Capsule twice daily (one
dose on Days 1 and 8 , twice-daily doses Days 2–7). In the
food-effect part, eight subjects received 200 mg FP-025
ASD-in-Capsule as a single dose in a randomized manner,
once under fasting conditions (treatment D) and once after
the intake of a high-fat breakfast (treatment E, following
Assessed for eligibility (n=88)
Excluded (n=15)
Not meeting inclusion criteria (n=14)
Declined to participate (n=1)
Approved screening but not included (n=9)
API-in-Capsule (n=18)
ASD-in-Capsule (n=30)
Randomized (n= 64)
Placebo
(n=16)
Cohort 8
450 mg
(n=6)
Cohort 7
350mg
(n=6)
Cohort 6
200mg
(n=6)
Cohort 5
100mg
(n=6)
Cohort 4
50 mg
(n=6)
Cohort 3
800mg
(n=6)
Cohort2
400mg
(n=6)
Cohort 1
200mg
(n=6)
Completed
(n=6)
Completed
(n-6)
Completed
(n=6)
Completed
(n=6)
Completed
(n=6)
Completed
(n=6)
Completed
(n=6)
Completed
(n=6)
Completed
(n-16)
Fig. 2 Subject flow diagram (single ascending dose, Study I). API active pharmaceutical ingredient, ASD amorphous solid dispersion
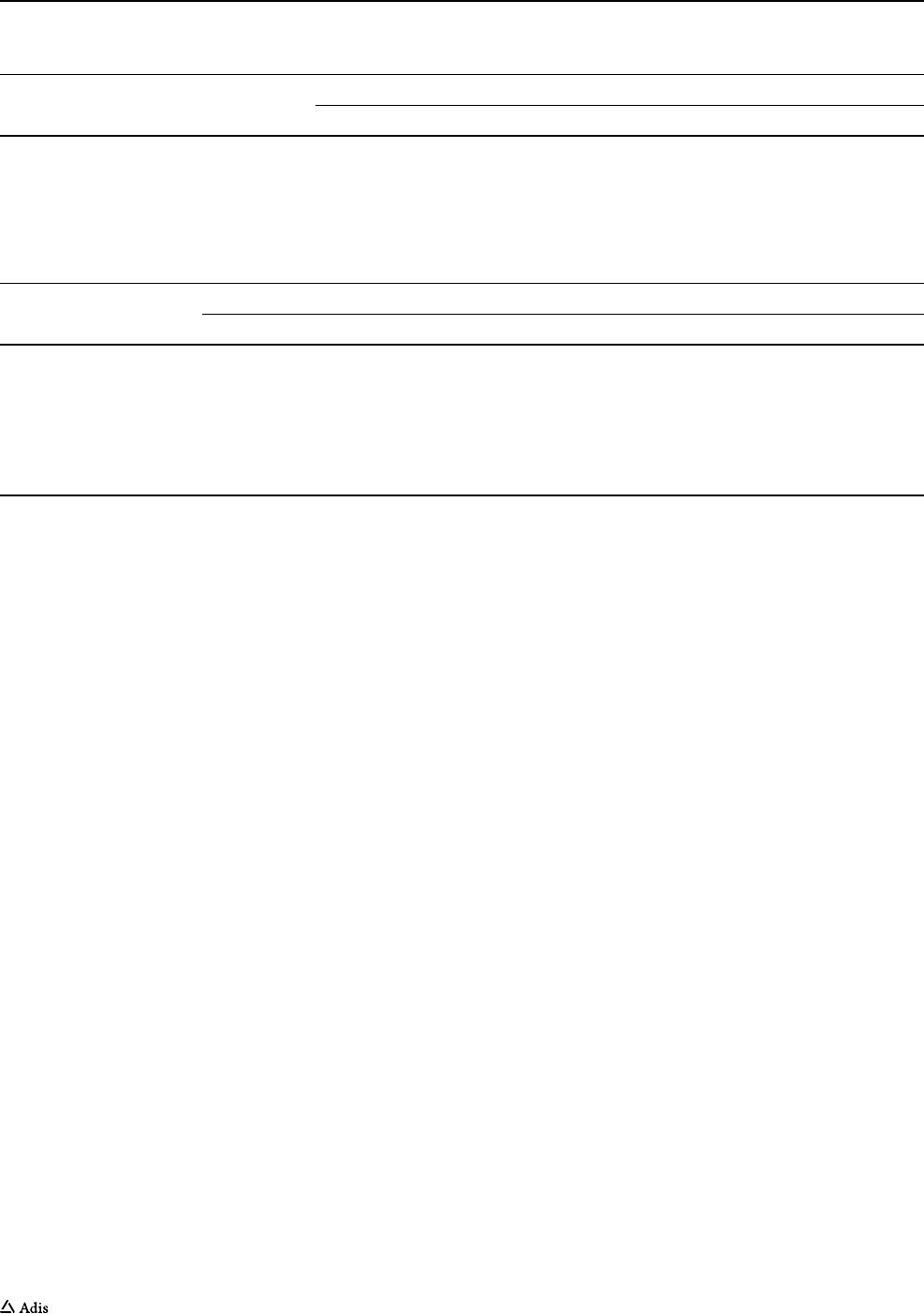
Fig. 3 Subject flow diagram
(multiple ascending-dose and
food-effect part, Study II)
Assessed for eligibility (n=76)
Excluded (n=25)
Approved screening but not included (n=19)
Multiple ascending dose
Food effect part
Randomized (n= 32)
Sequence ED
(Fed-Fasted)
200 mg
n=4
Sequence DE
(Fasted-Fed)
200 mg
n=4
Placebo
(n=6)
Cohort C
400 mg
(n=6)
Cohort B
200 mg
(n=6)
Cohort A
100 mg
(n=6)
Completed
(n=4)
Completed
(n=4)
Completed
(n=6)
Completed
(n=6)
Completed
(n=6)
Completed
(n=6)

Safety, Tolerability, and Pharmacokinetics of FP-025
the US Food and Drug Administration (FDA) composition),
with a wash-out period of 1 week between successive dos-
ing (Fig.3).
In both studies, matching placebo capsules were used.
The randomization code and randomization list were gener-
ated using SAS program version 9.3.
2.4 Safety andTolerability Assessments
andAnalysis
Safety and tolerability were assessed throughout the studies
by AE reporting, vital sign measurements, physical exami-
nation, laboratory tests, and ECG examinations. All adverse
events were coded according to MedDRA version 20.0.
2.5 Pharmacokinetic Assessments andAnalysis
In Study I, blood samples (6 mL/sample) for plasma concen-
trations of FP-025 were drawn at 0.5, 1, 2, 3, 4, 6, 8, 12, 16,
20, 24, and 36 h after each single dose. In Study II, multiple
blood samples were collected post-dosing on Day 1 (at 0.5,
1, 2, 3, 4, 6, 8, 12, and 16 h) and Day 8 (at 0.5, 1, 2, 3, 4, 6, 8,
10, 12, 16, 20, 24, 30, 36, and 48 h) as well as immediately
pre-morning dose on Days 2–7 and 2 h post-morning dose
on Days 4 and 6.
After collection, blood samples were kept in ice, and
within 15 min of collection were centrifuged at 2000 rpm for
10 min at 4°C, and, subsequently, the plasma was aliquoted,
frozen, and stored at –70°C pending pharmacokinetics
analyses. The samples were subjected to liquid chromatogra-
phy followed by analyses by tandem mass spectrometry (LC-
MS/MS). The analysis method used for the determination of
FP-025 in human plasma is a liquid-liquid extraction (LLE)
and LC-MS/MS. The LC-MS/MS method has two mobile
phases (mobile phase A and mobile phase B). The m/z tran-
sitions from Q1 mass (393.1 amu) to Q3 mass (194.7 amu)
was in 200 ms. The retention time of the component is 1.26
(min). Precision and accuracy of FP-025 plasma concentra-
tions were evaluated intraday, in three separate analysis runs
acquired on three different days, and interday, over the same
three analysis runs. This method shows adequate precision
and accuracy over a concentration range of 5.00 to 5000 ng/
mL. The assay is selective for FP-025. Stability of FP-025
in sample extracts was confirmed for 96 h at 2–8°C, and for
a batch size at the length injection of 160 extracts. Recov-
ery of FP-025 and the Internal Standard (IS) was consistent
over the concentration range, and the IS corrected well in
the recovery assessment. No carry-over or matrix effect was
observed for FP-025.
The plasma pharmacokinetics variables were derived by
non-compartmental analyses using Phoenix
®
WinNonlin
®
version 6.3 or higher (Pharsight Corporation Inc., Mountain
View, CA, USA).
Fecal excretion (> 97%) was the dominant elimination
route for 14C-FP-025-derived radioactivity after oral admin-
istration in rats, compared to ∼2% urinary recovery (unpub-
lished data), therefore no urine was collected.
No formal dose-proportionality assessments were done
for API-in-Capsule formulation, due to the low exposure.
For the ASD-in-Capsule formulation, the formal dose-pro-
portionality assessment was assessed using a power model
in which log(PK)=α+βXlog(dose). The area under the
plasma concentration-time curve from time zero to 12 h post
dose (AUC
0-12
) was used for the accumulation index. The
observed accumulation ratio calculated as mean AUC
0-12
(Day 8)/AUC
0-12
(Day 1) were 1.68, 1.68, and 1.69 for doses
of 100, 200, and 400 mg, respectively. At all doses, FP-025
accumulation was approximately 1.7-fold after twice-daily
administration.
2.6 Statistical Analysis
Safety and tolerability data were evaluated for the treated
population (all participants who took at least one dose of
study medications). Pharmacokinetic analyses were based
on data from treated participants in whom at least one phar-
macokinetic variable could be calculated and who did not
have any protocol violations that could interfere with these
evaluations. Descriptive statistics were used to evaluate the
safety, tolerability data, plasma concentrations, and pharma-
cokinetic variables of FP-025. Pharmacokinetic variables
were summarized using arithmetic mean, standard devia-
tion, median, minimum, maximum, the percent coefficient of
variation, geometric mean, and a two-sided 95% confidence
limit of the arithmetic mean and the geometric mean. Formal
calculations of sample sizes were not performed. Based on
the nature of the descriptive studies, the number of partici-
pants enrolled in each cohort was considered sufficient to
meet the objectives of these phase I studies and to allow for
assessments of pharmacokinetic variables.
For the evaluations of food effect on the pharmacokinetic
profile of FP-025, the analysis of variance (ANOVA) model
included sequence, treatment, and period as fixed effects and
subject nested within sequence as a random effect using the
SAS® mixed model procedure. Each ANOVA included
calculation of least squares means (LSM), the difference
between LSM under fed condition (Test) over fasted condi-
tion (Reference), and the standard error associated with this
difference.
At each time-point, mean, median, standard deviation,
minimum, maximum, number of available observations,
and change from baseline were summarized by numeric
parameters.

K.Abd-Elaziz et al.
3 Results
3.1 Subject Disposition andBlinding
In Study I, 88 male subjects were screened and 64 were
randomized (Fig.2). In Study II, 76 male and female sub-
jects were screened, 24 of whom were randomized in the
multiple-dose part, while eight subjects were randomized
in the food-effect part of the study (Fig.3).
All subjects completed the study. Overall, subjects at the
different dose levels were similar with regard to age, weight,
height, and body mass index (BMI) distribution (Tables1
and 2).
3.2 Safety andTolerability
All recorded AEs were self-limiting and considered to
be of mild intensity. No moderate, severe, or serious AEs
were reported. No clinically relevant changes from baseline
assessments were observed in weight, vital signs, physical
examination, laboratory, or ECG data after FP-025 at any
time point.
Only one possibly related AE was reported during the
SAD study (Table3). This AE, mild diarrhea, was expe-
rienced by one subject following the API-in-Capsule 800
mg dosing and recovered spontaneously before the follow-
up visit. In the MAD study, a total of seven AEs possibly
related to treatment with FP-025 were reported (Table3).
Two of these events, comprising fatigue and cough, were
reported by two subjects from the 100 mg FP-025 group.
The other five events were reported by three subjects from
the 200 mg FP-025 group, i.e., one subject with two episodes
of eye irritation, and erythema, one subject with dizziness,
and one subject with skin rash.
In the food-effect part of the study, only two AEs pos-
sibly related to treatment with FP-025 were reported in one
subject. Both events were headache, which occurred under
both fed and fasted conditions.
3.3 Pharmacokinetics: Single Ascending‑Dose
Study (Study I)
In the API-in-Capsule cohorts, median time to reach maxi-
mum plasma concentration (T
max
) was similar for FP-025
200 mg and 400 mg doses, i.e., 5 and 6 h, respectively,
whereas median T
max
appeared shorter (3.5 h) for the 800
mg dose. Mean maximum observed plasma concentration
(C
max
), area under the plasma concentration-time curve
from time zero to time t of the last measured concentration
(AUC
0–t
), and area under the plasma concentration-time
curve from time zero to infinity (AUC
0–inf
) increased in a
more than dose -proportional manner over the dose range of
200–400 mg, but decreased with the further doses (Table4).
In general, with the API-in-Capsule formulation, the expo-
sure to FP-025 was low and inconsistent, with substantial
inter-subject variability, expressed as percent coefficient of
variation of approximately 40%.
In the ASD-in-Capsule cohorts, median T
max
ranged from
1 to 2.5 h over the dose range of 50–450 mg after intake of
single doses. Mean C
max
, AUC
0–t
, and AUC
0–inf
increased in
a more than dose proportional manner over the dose range
between 50 and 100 mg. The drug exposure (mean C
max
,
AUC
0–t
, and AUC
0–inf
) increased in an approximately dose-
proportional manner from 100 to 450 mg after intake of
single doses of FP-025 (Table4). Dose-proportionality was
found for C
max
, AUC
0–t
, and AUC
0–inf
over the dose range
Table 1 Demographic and baseline characteristics of subjects participating in the single ascending-dose study (Study I)
Data are presented in mean (standard deviation) unless otherwise specified
API active pharmaceutical ingredient, ASD amorphous solid dispersion, SD standard deviation, n number, cm centimeter, kg kilogram, m meter,
BMI body mass index
Variable Placebo
(SD)
API-in-
capsule 200
mg (SD)
API-in-
capsule 400
mg (SD)
API-in-
capsule 800
mg (SD)
ASD-in-
capsule 50
mg (SD)
ASD-in-
capsule 100
mg (SD)
ASD-in-
capsule 200
mg (SD)
ASD-in-
capsule 300
mg (SD)
ASD-in-
capsule 450
mg (SD)
Subject, n 16 6 6 6 6 6 6 6 6
Age, years 28.8 (6.28) 26.3 (2.66) 28.5 (6.69) 29.5 (8.34) 28.5 (2.51) 28.0 (4.47) 28.3 (5.57) 31.0 (5.44) 25.8 (2.79)
Male, n 16 6 6 6 6 6 6 6 6
Asian race,
n
16 6 6 6 6 6 6 6 6
Height, cm 173.19
(5.82)
170.95
(6.85)
174.33
(8.98)
173.83
(5.38)
175.77
(8.94)
176.27
(5.79)
177.58
(12.05)
173.5 (5.13) 169.0 (4.34)
Weight, kg 70.7 (9.72) 63.83 (7.89) 64.67 (7.0) 69.17 (7.86) 74.53
(16.22)
71.5 (8.09) 73.2 (14.46) 69.2 (6.56) 62.83 (5.23)
BMI, kg/m
2
23.50 (2.35) 21.88 (2.96) 21.25 (1.25) 22.95 (2.96) 23.97 (2.29) 23.00 (2.29) 23.17 (3.46) 23.00 (1.94) 21.97 (1.10)

Safety, Tolerability, and Pharmacokinetics of FP-025
of 100–450 mg after single doses of FP-025. Mean values
of T
1/2
were independent of the dose administered over the
range of 50–450 mg after single oral doses of FP-025. Mean
apparent total clearance (CL/F) and mean apparent volume
of distribution (V
z
/F) of FP-025 were independent of the
dose administered over the range of 100–300 mg. The high-
est exposure was observed in the 450 mg group (Table4,
Fig.4). Compared to the API-in-Capsule formulation,
FP-025 administration using the ASD-in-Capule formula-
tion achieved higher and more consistent exposure, as well
as lower inter-subject variability.
3.4 Pharmacokinetics: Multiple Ascending‑Dose
Study andFood‑Effect Part (Study II)
The peak exposure (C
max
) and the systemic exposure
(AUC) to FP-025 increased in a more-than-dose propor-
tional manner from 100 to 200 mg and approximately in
a dose-proportional manner from 200 to 400 mg after
multiple doses for a week. Median T
max
of FP-025 was
similar among the three escalating dose levels on both
Day 1 and Day 8 (Fig.5, Table5). T
1/2
of FP-025 at sin-
gle doses of 100, 200, and 400 mg was 6.9, 6.0, and 6.5
h on Day 1, respectively; the corresponding values after
intake of multiple doses was 8.4, 8.1, and 6.6 h on Day 8,
respectively. CL/F was 28.7, 20.3, and 24.0 L/h after sin-
gle doses of FP-025 at 100, 200, and 400 mg, respectively.
The corresponding values after multiple doses were 23.1,
16.2, and 18.2 L/h, respectively. The mean trough (pre-
dose) plasma concentrations were used to assess the time
to steady state. Steady state was achieved by Day 6 for all
three dose levels. The AUC ratio of FP-025 (Day 8/Day 1)
was approximately 1.7-fold after twice-daily administra-
tion at all three dose levels.
Co-administering FP-025 with food appears to have
no effect on overall exposure (similar AUCs were
Table 2 Demographic and baseline characteristics of subjects participating in the multiple ascending dose study including the food effect part
Data are presented in mean (standard deviation) unless otherwise specified
API active pharmaceutical ingredient, ASD amorphous solid dispersion, SD standard deviation, n number, cm centimeter, kg kilogram, m
meter,BMI body mass index
Treatment A = oral dose of 100 mg; treatment B = oral dose of 200 mg; treatment C = oral dose of 400 mg. Treatments A, B, and C are ASD-
in-Capsule. Treatment D = single oral dose of 200 mg ASD-in-Capsule in fasted condition and treatment E = single oral dose of 200 mg ASD-
in-Capsule in fed condition after intake of a high-fat, high-calorie breakfast (following the US Food and Drug Administration composition)
Variable Placebo (SD) Treatment A (SD) Treatment B (SD) Treatment C (SD) Treatment D/E (SD)
Subject, n 6 6 6 6 8
Age, years 30.5 (13.02) 32.3 (10.82) 34.0 (11.1) 33.2 (14.52) 44.5 (12.4)
Male, n 6 6 6 5 7
Black race, n 0 2 3 0 0
White race, n 6 4 3 6 8
Height, cm 183.17 (9.96) 181.73 (4.36) 181.05 (7.95) 182.85 (7.78) 183.78 (7.09)
Weight, kg 82.87 (13.52) 81.95 (9.48) 69.4 (12.12) 73.93 (7.9) 85.33 (13.89)
BMI, kg/m
2
24.6 (2.37) 24.93 (3.70) 21.12 (3.01) 22.18 (1.96) 25.15 (2.95)
Table 3 Adverse events possibly related to drug administration reported during both single ascending-dose (Study I) and multiple ascending-
dose including the food effect (Study II) studies
API active pharmaceutical ingredient, ASD amorphous solid dispersion
Adverse events description Study Dose level (mg) FP-025 formulation Outcome
Diarrhea (n=1) Single ascending dose 800 API Resolved
Fatigue (n=1) Multiple ascending dose 100 ASD Resolved
Cough (n=1) Multiple ascending dose 100 ASD Resolved
Eye irritation (n=1) Multiple ascending dose 200 ASD Resolved
Erythema (n=1) Multiple ascending dose 200 ASD Resolved
Dizziness (n=1) Multiple ascending dose 200 ASD Resolved
Skin rash (n=1) Multiple ascending dose 200 ASD Resolved
Headache (n=1) Food effect fed status 200 ASD Resolved
Headache (n=1) Food effect fasting status 200 ASD Resolved
K.Abd-Elaziz et al.
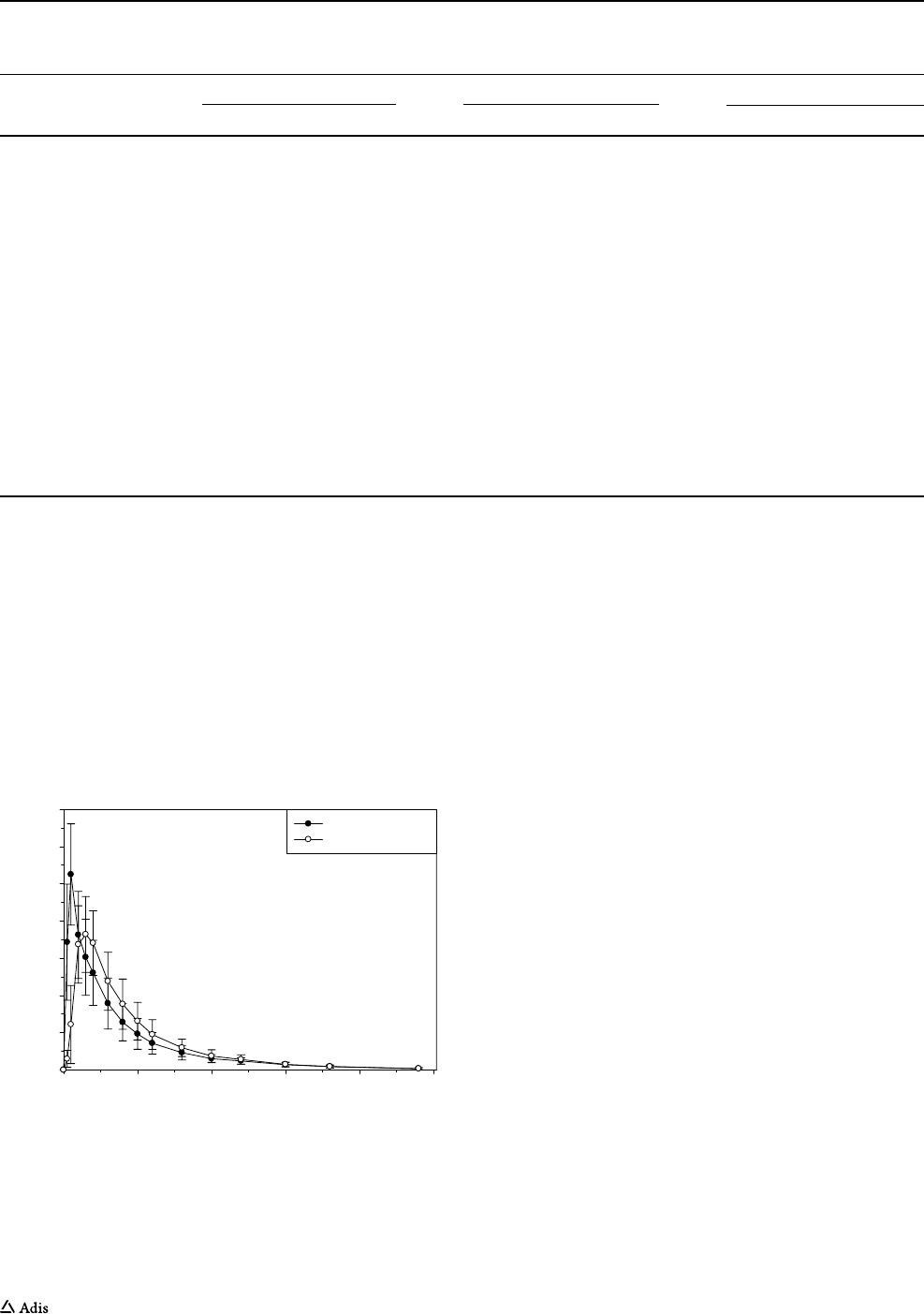
obtained with or without food), although the peak time
increased from 1 to 2.5 h and a lower peak concentra-
tion was observed (1050 ng/mL vs. 787 ng/mL) (Fig.6).
T
1/2
was 9.4 and 8.4 h under fasted and fed conditions,
respectively.
4 Discussion
The present paper reports the first-in-man data of a
novel, highly selective, oral MMP-12 inhibitor, FP-025,
from two phase I (SAD, MAD) studies conducted in 96
healthy subjects. FP-025 exposure appears substantially
increased with an ASD-based formulation, which has been
commonly used to improve oral absorption of drugs with
low solubility [39]. We also found that the ASD-in-Cap-
sule formulation resulted in shorter peak time, increased
peak concentrations, and higher overall plasma exposure.
Moreover, inter-subject variability of the pharmacokinet-
ics variables of FP-025 was lower in the ASD-in-Capsule
groups, and an overall dose-proportional increase was
observed in the majority of the dose range tested. For
example, at the 200 mg FP-025 dose, the ASD-in-Capsule
formulation resulted in 30-fold higher C
max
and AUC
0–inf
when compared to the API-in-Capsule formulation. The
percent coefficient of variation of these parameters also
declined from approximately 30–40% to less than 30%.
The decrease in inter-subject variability was more appar-
ent at higher doses. These observations are consistent with
those of many marketed products using this technology to
improve oral bioavailability of drugs with low solubility
[40].
Although the administration of FP-025 with a high-fat,
high-calorie diet appears to result in a shift in the plasma
concentration profile of the drug, the overall exposure
remains relatively unchanged, suggesting a minor delay in
rate of absorption, but without an effect in extent of absorp-
tion or bioavailability.
Because FP-025 is developed for chronic disease treat-
ment, the minor shift in pharmacokinetic profile may not be
significant to mandate dosing with or without food.
Across all dose levels and dosing regimens that have been
studied, FP-025 was safe and generally well tolerated. There
were only a few AEs that were mild, short-lasting, and self-
limited. There was no increase in frequency or intensity of
AEs at higher dose levels. Our data extend and complement
the few clinical studies with previously tested, often non-
selective MMP inhibitors.
Table 4 FP-025 pharmacokinetic parameters (mean and SD) after a single oral dose of FP-025 API-in-Capsule formulation at 200, 400, and 800
mg and FP-025 ASD-in-Capsule formulation at 50, 100, 200, 300, and 450 mg in Study I
Data are arithmetic mean (standard deviation) except for T
max
, which is reported as median (range)
The standard deviation was not calculated for some parameters because the total number was less than three
API active pharmaceutical ingredient, ASD amorphous solid dispersion AUC
0–t
area under the plasma concentration-time curve from time zero
to time t of the last measurable concentration, AUC
0–inf
area under the plasma concentration-time curve from time zero to infinity, C
max
maxi-
mum observed plasma concentration, T
1/2
apparent terminal elimination half-life, T
max
time to reach maximum plasma concentration, λz terminal
elimination rate constant, a* median
Parameter FP-025 API-in-Capsule
200 mg 400 mg 800mg
C
max
(ng/mL) 34.1 (10.7) 90.6 (40.2) 63.6 (93.2)
T
max
(h) 5 (2–6) 6 (4–12) 3.5 (3–12)
AUC
0–t
(ng·h/mL) 448 (196) 1380 (771) 1040 (630)
AUC
0–inf
(ng·h/mL) 313 1480 (528) 935
λz (1/h) 0.103 0.0748 (0.01) 0.0569
T
1/2
(h) 6.7 9.4 (1.5) 12.5
FP-025 ASD-in-Capsule
50 mg 100 mg 200 mg 300 mg 450 mg
C
max
(ng/mL) 193 (25.2) 498 (113) 1000 (269) 1580 (309) 2570(270)
T
max
(h) 2 (1–3) 1 (1–3) 2.5 (1–3) 1 (1–3) 2 (1–6)
AUC
0–t
(ng·h/mL) 1580 (402) 4520 (1160) 8960 (2540) 12,900 (2260) 25,400 (4980)
AUC
0–inf
(ng·h/mL) 1680 (472) 4720 (1260) 9180 (2630) 13,200 (2390) 26,000 (5370)
λz (1/h) 0.137 (0.06) 0.113 (0.04) 0.107 (0.03) 0.108 (0.02) 0.108 (0.02)
T
1/2
(h) 6.2 (3.2) 6.7 (2.0) 6.9 (1.9) 6.7 (1.6) 6.7 (1.4)
Safety, Tolerability, and Pharmacokinetics of FP-025
Early MMP-12 inhibitors showed protective and/or
anti-inflammatory efficacy in several animal models of
inflammatory airway disease underscoring the relevance
of MMP-12 targeting [31, 32, 41–43]. Chung and col-
leagues reported on the anti-inflammatory efficacy of
AZ11557272, an MMP-12 inhibitor, in a guinea-pig model
of COPD completely eradicating inflammatory cells and
desmosine in bronchoalveolar lavage (BAL) fluid [44].
Likewise, Le Quément and colleagues showed inhibition
of MMP-12 with AS111793, another selective MMP-12
inhibitor, on cigarette smoke-induced airway inflammation
in a mouse model [43].
Based on their antiangiogenic and putative antimeta-
static properties, several non-selective MMP-inhibitors
have been developed and tested in patients with cancer.
BMS-275291 (rebimastat), a non-selective MMP inhibitor
(targeting MMPs 1, 2, 8, 9, and 14) with limited efficacy,
was associated with unexpected toxicity (i.e., severe sys-
temic side effects, including fatigue, allergic reactions, and
arthralgia) resulting in early termination of phase ll and
phase lll clinical trials [19–21]. BAY 12-9566 (tanomastat),
another non-selective MMP inhibitor (targeting MMPs 2, 3
and 9), failed to show consistent clinical efficacy [22, 23].
More recently, BB-2516 (marimastat), a broad-spectrum
MMP inhibitor, had ameliorated liver injuries and had lim-
ited inflammation processes but caused liver fibrosis in the
mouse model of repeated carbon tetrachloride administra-
tion [45].
So far, there are only limited human data on selective
MMP inhibitors. AZD1236, an oral dual MMP-9/12 inhibi-
tor, was evaluated in two phase II studies in patients with
moderate to severe COPD. During 6 weeks, AZD1236
showed an overall good safety and tolerability [24, 25].
There was no apparent efficacy on any of the exploratory
clinical outcomes (i.e., Clinical COPD Questionnaire
(CCQ) scores, lung function, 6-min walk test (6MWT),
body mass index, obstruction, dysponea and excercise
(BODE) index) was observed, despite a potential signal of
efficacy reflected by a reduction in urinary desmosine excre-
tion [24, 25].
In vitro studies with FP-025 confirmed its high affinity to
inhibit MMP-12 with a 90-fold selectivity over the next clos-
est family member , indicative of a high selectivity, which
Time (h)
010203
04
0
Concentration (ng/mL)
0
20
40
60
80
100
120
140
API-in-Capsule, 200 mg (N=6)
API-in-Capsule, 400 mg (N=6)
API-in-Capsule, 800 mg (N=6)
Time (h)
010203
04
0
Concentration (ng/mL)
0
500
1000
1500
2000
2500
3000
ASD-in-Capsule, 50 mg (N=6)
ASD-in-Capsule, 100 mg (N=6)
ASD-in-Capsule, 200 mg (N=6)
ASD-in-Capsule, 300 mg (N=6)
ASD-in-Capsule, 450 mg (N=6)
Fig. 4 Mean (±SD) plasma FP-025 concentration versus time plots
after administration of single oral doses of 200 mg, 400 mg, and 800
mg API-in-Capsule and 50 mg, 100 mg, 200 mg, 300 mg, and 450
mg ASD-in-Capsule in fasted condition. API active pharmaceutical
ingredient, ASD amorphous solid dispersion
DAY 1
Time (h)
0510 15 20 25
Concentration (ng/mL)
0
500
1000
1500
2000
2500
3000
3500
Treatment A (N=6)
Treatment B (N=6)
Treatment C (N=6)
DAY 8
Time (h)
01020304
05
0
Concentration (ng/mL)
0
1000
2000
3000
4000
5000
Treatment A (N=6)
Treatment B (N=6)
Treatment C (N=6)
Fig. 5 Mean (± SD) plasma FP-025 concentration versus time plots
after administration of multiple oral doses of 100 (treatment A, n =
6), 200 (treatment B, n = 6) and 400 (treatment C, n = 6) mg ASD-
in-Capsule on day 1 and on day 8 of Study II. ASD amorphous solid
dispersion
K.Abd-Elaziz et al.
has been shown to translate into a good safety profile. Com-
bined with a favorable pharmacokinetic profile, these prop-
erties will enable FP-025 to fill the gap in selective and safe
MMP inhibitors and warrant further investigation of efficacy
in relevant patient populations including asthma, COPD and,
pulmonary fibrosis.
5 Conclusions
FP-025, an oral MMP-12 inhibitor, was safe, well tolerated,
and showed a favorable pharmacokinetic profile in healthy
subjects at all single and multiple ascending doses tested in
the ASD-in-Capsule formulation. Results from these studies
warrant exploration and further research of its activity and
therapeutic potentials in target populations.
Acknowledgements The authors would like to acknowledge dr. Rene
Lutter (Department of Respiratory Medicine and Experimental Immu-
nology Amsterdam University Medical Center, University of Amster-
dam, Amsterdam, the Netherlands) for his critical review of the manu-
script; Chern-En Chiang and Diahn-Warng Perng (General Clinical
Research Center, Taipei Veterans General Hospital, Taipei, Taiwan and
School of Medicine, National Yang-Ming University, Taipei, Taiwan)
for their assistance in execution of Study I; and Trista Lin and Yu-Fan
Chen (QPS-Qualitix , Taipei, Taiwan) for their assistance in biostatisti-
cal analysis of safety and efficacy data of both studies.
Declarations
Funding Both studies were funded by Foresee Pharmaceuticals Co.,
Ltd. Taipei, Taiwan.
Table 5 Summary statistics of pharmacokinetic parameters of FP-025 after multiple ascending oral doses of treatments A (100 mg ASD-in-
Capsule), B (200 mg ASD-in-Capsule), and C (400 mg ASD-in-Capsule) in healthy male and female subjects in Study II
ASD amorphous solid dispersion AUC
0-t
area under the plasma concentration-time curve from time zero to time t of the last measurable concen-
tration, AUC
0-inf
area under the plasma concentration-time curve from time zero to infinity, C
max
maximum observed plasma concentration, T
1/2
apparent terminal elimination half-life, T
max
time to reach maximum plasma concentration, λz terminal elimination rate constant, Mean arithme-
tic mean, SD standard deviation
a
Median
Parameter Treatment A Treatment B Treatment C
N Mean (SD) N Mean (SD) N Mean (SD)
Day 1
C
max
(ng/mL) 6 448 (65.9) 6 1200 (338) 6 2730 (671)
T
max
(h)
a
6 1.00 (0.50-2.00) 6 1.02 (1.00–4.00) 6 1.00 (1.00–3.00)
AUC
0–12
(ng·h/mL) 6 2670 (532) 6 7810 (1870) 6 14,600 (4480)
AUC
0–24
(ng·h/mL) 6 3290 (593) 6 9720 (2320) 6 18,200 (6520)
AUC
0–t
(ng·h/mL) 6 3290 (593) 6 9710 (2320) 6 18,200 (6510)
AUC
0–inf
(ng·h/mL) 6 3550 (547) 6 10,400 (2520) 6 19,900 (7950)
T
1/2
(h) 6 6.9 (1.8) 6 6.0 (1.0) 6 6.5 (1.9)
Day 8
C
max
(ng/mL) 6 624 (141) 6 1920 (366) 6 3710 (932)
T
max
(h)
a
6 2.00 (1.00-3.00) 6 1.50 (1.00–3.00) 6 1.50 (0.50–2.00)
C
min
(ng/mL) 6 156 (27.4) 6 524 (196) 6 1010 (651)
AUC
0–12
(ng·h/mL) 6 4380 (543) 6 13,100 (3440) 6 24,700 (8350)
AUC
0–inf
(ng·h/mL) 6 6180 (889) 6 19,200 (5970) 6 36,300 (17,900)
T
1/2
(h) 6 8.4 (2.6) 6 8.1 (2.1) 6 6.6 (2.3)
Time (h)
01020304
05
0
Concentration (ng/mL)
0
200
400
600
800
1000
1200
1400
Treatment D (N=8)
Treatment E (N=8)
Fig. 6 Mean (± SD) plasma FP-025 concentration versus time plots
after administration of single oral doses of 200 mg ASD-in-Capsule
in fasted condition (treatment D, n = 8) and 200 mg ASD-in-Cap-
sule after intake of a high-fat, high-calorie breakfast, following the
US Food and Drug Administration composition (treatment E, n = 8).
ASD amorphous solid dispersion
