
ORIGINAL PAPER
Bimodal evolution of the killer cell Ig-like receptor (KIR)
family in New World primates
Luis F. Cadavid & Catalina Palacios & Juan S. Lugo
Received: 26 April 2013 /Accepted: 26 June 2013
#
Springer-Verlag Berlin Heidelberg 2013
Abstract The immunoglobulin-like receptor (KIR) gene
family in New World primates (Platyrrhini) has been
characterized only in the owl monkey (Aotus sp.). To gain
a better understanding of the KIR system in Platyrrhini, we
analyzed a KIR haplotype in Ateles geoffroyi, and sequenced
KIR complementary DNAs (cDNAs) from other three
Atelidae species, Ateles hybridus, Ateles belzebuth,and
Lagothrix lagotricha. Atelidae expressed a variable set of
activating and inhibitory KIRs that diversified independently
from their Catarrhini counterparts. They had a unique mech-
anism to generate activating receptors from inhibitory ones,
involving a single nucleotide deletion in exon 7 and a change
in the donor splice site of intron 7. The A. geoffroyi haplotype
contained at least six gene models including a pseudogene,
two coding inhibitory receptors, and three coding activating
receptors. The centromeric region was in a tail-to-tail orienta-
tion with respect to the telomeric region. The owl monkey
KIR haplotype shared this organization, and in phylogenetic
trees, the centromeric genes clustered together with those of A.
geoffroyi, whereas their telomeric genes clustered independent-
ly. KIR cDNAs from the other Atelidae species conformed to
this pattern. Signatures of positive selection were found in
residues predicted to interact with the major histocompatibility
complex. Such signatures, however, primarily explained vari-
ability between paralogous genes but not between alleles in a
locus. Atelidae, therefore, has expanded the KIR family in a
bimodal fashion, where an inverted centromeric region has
remained relatively conserved and the telomeric region has
diversified by a rapid process of gene duplication and diver-
gence, likely favored by positive selection for ligand binding.
Keywords KIR
.
Killer cell Ig-like receptor
.
New World
primates
.
Atelidae
Introduction
Natural killer (NK) cells are granular lymphocytes with
cytotoxic effects against virus-infected, allogeneic, and
transformed cells (Moretta et al. 2003). Their function is
largely controlled by a variable set of activating and inhibi-
tory receptors specific for major histocompatibility complex
(MHC) class I molecules (McQueen and Parham 2002). An
altered expression of MHC class I molecules on the target
cell initiates an activating signal cascade leading to cytotox-
icity, whereas normal expression of MHC class I molecules
triggers an inhibitory signaling resulting in anergy (Cheent
and Khakoo 2009). NK cell receptors belong to two struc-
tural classes, the lectin-like and the immunoglobulin (Ig)-
like receptors (KIRs). The former are type II transmembrane
proteins represented by the CD94/NKG2 family and are
encoded in the natural killer complex on human chromo-
some 12p13 (Ryan and Seaman 1997). The latter are type I
transmembrane proteins encoded in the leukocyte receptor
complex (LRC) on human chromosome 19q13 and are typ-
ically constituted by two or three Ig domains, a stem region,
Nucleotide sequence data reported are available in the GenBank
database under accession numbers KF011940-KF011974.
Electronic supplementary material The online version of this article
(doi:10.1007/s00251-013-0719-4) contains supplementary material,
which is available to authorized users.
L. F. Cadavid (*)
:
C. Palacios
:
J. S. Lugo
Department of Biology and Institute of Genetics, Universidad
Nacional de Colombia, Cr. 30 no. 45-08, Bogotá, Colombia
e-mail: [email protected]
Present Address:
C. Palacios
Department of Biological Sciences, Universidad de los Andes,
Bogotá, Colombia
Immunogenetics
DOI 10.1007/s00251-013-0719-4

a transmembrane domain, and a short or long cytoplasmic
domain (Barrow and Trowsdale 2008). KIRs with long cy-
toplasmic domains function as inhibitory receptors due to the
presence of two immunoreceptor tyrosin-based inhibitory
motifs (ITIMs), whereas KIRs with short cytoplasmic do-
mains function as activating receptors due to the presence of
a positively charged residue at the transmembrane domain
allowing the interaction with DAP12 molecules, which
posses immunoreceptor tyrosin-based activating motifs (La-
nier 2003).
The human KIR gene region on the LRC is flanked by the
leukocyte Ig-like receptor (LILR) cluster at the centromeric
end and by the IgA receptor gene (FcAR) at the telomeric end
(Barrow and Trowsdale 2008). The gene content of the KIR
family varie s between individuals, where most variation
occurs in two regions delimitated by conserved genes located
at the centromeric end (KIR3DL3), central region (KIR3DP1
and KIR2DL4), and telomeric end (KIR3DL2) of the cluster
(Parham and Moffett 2013). The 13 genes and 2 pseudogenes
identified in the human KIR region are grouped into two types
of haplotypes (A and B). Haplotype A mainly contains genes
encoding inhibitory receptors, whereas haplotype B primarily
contains genes encoding activating receptors (Parham and
Moffett 2013). This KIR cluster organization has been rela-
tively well conserved in Catarrhini primates (humans, apes,
and Old World monkeys) with species-specific differences
concentrated in the two variable regions (Palacios et al.
2011;Parhametal.2010). Catarrhini KIRs belong to four
evolutionary lineages that differ in their structure and speci-
ficity to MHC class I molecules (Guethlein et al. 2007b;
Blokhuis et al. 2011). Lineage I KIRs are represented by the
human KIR2DL4 and recognize the nonclassical MHC class I
molecule MHC-G. Lineage II KIRs are represented by the
human KIR3DL1 and KIR3DL2 and recognize MHC-A and
MHC-B molecules, whereas lineage III KIRs (i.e., KIR2DL2
and KIR2DS1) recognize MHC-C and certain MHC-B allo-
types. Finally, lineage V is represented by the human KIR3DL3
with no known MHC specificity.
In contrast with the solid understanding of the KIR system
in Catarrhini, KIR genes in Platyrrhini (New World pri-
mates) have only been reported in one species, the owl
monkey (Aotus sp.) (Cadavid and Lun 2009). Genomic
analyses in this species showed that the owl monkey KIR
region is flanked by the LILR and FcAR genes and contain at
least seven genes encoding putative activating and inhibitory
receptors with three or four Ig domains. Owl monkey KIRs
do not belong to any of the Catarrhini evolutionary lineages,
constituting an independent lineage VI with no apparent
direct orthology to any of the genes from the other four
lineages. In order to have a better understanding of the compo-
sition and diversification patterns of the KIR system in
Platyrrhini, we have analyzed the KIR homologous genomic
region in the black-handed spider monkey (Ateles geoffr oyi),
and have cloned and sequenced KIR cDNAs from other three
Atelidae species, the brown spider monkey (Ateles hybridus), the
long-haired spider monkey (Ateles belzebuth), and the brown
woolly monkey (Lagothrix la gotricha). The Atelidae family in-
cludes five genera (Alouatta, Ateles, Brachyteles, Lagothrix, and
Oreonax) distributed along Central and South America, and has
the largest species of New World monkeys (Hershkovitz 1977).
Atelidae diverged from the Aotus species about 19.4±5. 0 million
years ago (MYA) (Porter et al.
1997) whereas Ateles and
Lagothrix genera diverged some 11. 8±1.2 MYA (Opazo et al.
2006). The diversification of the genus Ateles occurred about
1.1±0.2 MYA (Fabre et al. 2009).
Materials and methods
Genomic sequence of the Ateles geoffroyi KIR haplotype
A draft sequence of the bacterial artificial chromosome
(BAC) clone UC1-11A1 containing the black-handed spider
monkey (A. geoffroyi) KIR haplotype was obtained from
publicly available databases (GenBank accession number
AC234015). This BAC clone was sequenc ed by the NIH
Intramural Sequencing Center ( www.nisc. nih.gov) as part of
the Comparative Vertebrate Sequencing Initiative and the
sequence was released on February 18, 2009. The BAC
clone draft sequence was composed by 11 ordered contigs
separated by gaps of unknown size. In addition, to comple-
ment and refine the analysis of the owl monkey (Aotus sp.)
KIR locus previously reported using the BAC clone CH258-
187H8 (AC193069) (Cadavid and Lun 2009), the sequence
of a second BAC clone (CH258-69P6, AC234076) from this
species was obtained. This sequence was also generated by
the NIH Intramural Sequencing Center. Each of these contigs
was analyzed with the Genscan software (http://genes.mit.
edu/GENSCAN.html) to predict open readi ng frames and
exon–intron structure of gene models. The gene models were
subsequently compared against sequence databases by
BLAST (NCBI, http://www.ncbi.nlm.nih.gov) to identify
KIR genes. Intronic sequences were analyzed for retroelements
and repetitive elements using RepeatMasker (http://ftp.genome.
washington.edu/RM/Repeat-Masker .html).
Samples and PCR, cloning, and sequenc ing
Peripheral blood samples (0.5–1.0 ml) were obtained
from three brown spider monkeys (A. hybridus—individuals
110, 180, and 190), one long-haired spider monkey (A.
belzebuth—individual 170), and two brown woolly monkeys
(L. lagotricha—individuals 300 and 660). Animals were
maintained at two Colombian centers for rescue and rehabil-
itation of wild fauna (Corporación Autónoma Regional de
Norte de Santander and Corporación Autónoma Regional
Immunogenetics

del Alto Magdalena) under the animal care and use committee
policies of the respective institutions. Total RNA was isolated
with the Trizol reagent (Invitrogen) and used for complemen-
tary DNA (cDNA) synthesis with the Reverse Transcription
System kit (Fermentas). Full-length KIR cDNAs were
amplified by PCR with forward primer AteKIRPS-F
5′-ATGTCGCTCATGGTCRTCA GCATG-3′ and reverse
primer NWPKIRL1-R 5′-CTAGAGGACCCCTCAGGG-
3′, which anneal at KIR gene exons 1 and 9, respec-
tively. The reaction contained 400 μM dNTPs, 2 mM MgCl
2
,
and 0.5 U of Taq DNA polymerase (Invitrogen) in a volume of
25 μl. Amplification was carried out with an initial cycle of
5 min at 94 °C, followed by 35 cycles of 95 °C for 30 s, 60 °C
for 60 s, and 72 °C for 90 s, with a final extension of 72 °C for
10 min. PCR products were precipitated with 0.3 M sodium
acetate/ethanol, cloned into the pGEM-T Easy vector (Promega),
and at least 12 clones per individual were sequenced with the
Sanger method. Reported sequences derived from at least two
identical plasmid clones.
Sequence and evolutionary analyses
Sequences were aligned with the ClustalW software (Thomp-
son et al. 1994), and the alignment was manually edited in the
BioEdit sequence editor (www.mbio.ncsu.edu/BioEdit/bioedit.
html). Nucleotide distances were estimated with the Kimura
two-parameter model (Kimura 1980) with the variance estimat-
ed by the bootstrap method using the MEGA 5.05 package
(Tamura et al. 2011). Phylogenetic analyses were carried out
with the Neighbor-joining, maximum likelihood, and Bayesian
approaches based on a GTR+I+G nucleotide substitution mod-
el as indicated by the software ModelTest (Posada and Crandall
1998). Neighbor-joi ning and maximum likelihood phylogenies
were constructed with the MEGA 5.05 package (Tamura et al.
201 1), and their reliability was evaluated by bootstrap with
1,000 replicates, whereas Bayesian phylogenies were inferred
with MrBayes software (Huelsenbeck and Ronquist 2001;
Ronquist and Huelsenbeck 2003) using 10,000,000 iterations.
Natural selection acting on KIR genes was evaluated with the
test partitioning approach for robust inference of selection
(PARRIS), which uses likelihood ratio tests to identify the ratio
of nonsynonymous substitutions (dN) to synonymous substitu-
tions (dS) over the entire alignment (Scheffler et al. 2006 ), and
the test mixed effects model of evolution (MEME) which is a
phylogeny-based model that describe the evolution of individ-
ual codons in a tree branch by a continuous-time stationary
Markov process (Murrell et al. 2012). These two methods were
implemented in the datamonkey.org server. Finally, homology
modeling of Platyrrhini KIR structures was carried out with the
Swiss-Model suite (Arnold et al. 2006) using the human
KIR3DL1 structure (PDB 3VH8) as template. The models were
subjected to quality validation with the QMEAN score (Benkert
et al. 2011) implemented in the Swiss-Model suite and by
calculating the root mean square deviation (RMSD) from the
template with the VMD program (Humphrey et al. 1996).
Results
Organization of the KIR haplotype in Ateles geoffroyi
A. geoffroyi BAC clone UC1-11A1 (accession number
AC234015) contained six complete KIR gene models and
the FcAR framework genes in a single contig of 92,586 bp
(Fig. 1). The other KIR flanking gene, LILR, was in a
different contig of 12,226 bp separated from the KIR-con-
taining contig by a gap of unknown size. The intronic and
intergenic regions in the haplotype contained the typical
retroelements and repetitive elements (MLT1D/ LTR33A,
MSTB1, and MER70B) of primate KIRs located between
the LILR and FcAR genes (Sambrook et al. 2005). The first
two gene models at the centromeric end of the cluster, Atge-
KIR3DP1 and Atge-KIR3DL1, were in the opposite tran-
scription orientation with respec t to the other four gene
models (Fig. 1). The pseudogene Atge-KIR3DP1 lacked
exons 6–9 and had a single nucleotide deletion at exon 3
(position 131) that changed the reading frame to generate a
premature stop codon in the same exon. It also had a second
stop codon at exon 5 (position 702–704) due to a single
nucleotide substitution. The second gene model, Atge-
KIR3DL1, was predicted to encode a receptor with three Ig
domains and a long cytoplasmic tail with two putative ITIMs
(Supplementary Fig. 1). This gene model, however, lacked
exon 6 encoding for the stem domain, a featu re also observed
in KIRs from primate lineage V, represented by the human
KIR3DL3. The owl monkey (Aotus sp.) KIR haplotype
(Fig 1) contained also three gene models transcribed in the
opposite direction (Om-KIR3DP1 , Om-KIR3DL1, and Om-
KIR3DS1) (Cadavid and Lun 2009), indicating that an an-
cient inversion of the KIR centromeric region occurred in the
common ancestor of Ateli dae and Aotinae at least some
20 MYA. The next three gene models downstream in the A.
geoffroyi KIR cluster, Atge-KIR3DS1-3, were predicted to
encode activating receptors with three Ig domains, the pos-
itively charged residue arginine at the fourth position of the
transmembrane region, and a short cytoplasmic domain. The
truncated cytoplasmic domain was the result of a series of
mutations in exon 7 and intron 7 that included a single
nucleotide deletion three positions before the last nucleotide
of exon 7 that changed the reading frame, a G to A substitu-
tion at the first position of intron 7 that altered the donor
splicing site (GA^GTAAGT), and a single nucleotide dele-
tion at position 58 of intron 7 that changed further the
reading frame (Fig. 2). In addition, these models had a
putative secondary donor splice site (AG^GTGT) at posi-
tions 66–71 of intron 7, predicted at a 54 % of confidence
Immunogenetics

using the software GeneSplicer (Pertea et al. 2001). As a
consequence of these mutations, exon 7 was predicted to be
transcribed together with the first 65 nucleotides of intron 7,
and joined to exon 8 during messenger RNA processing in an
altered phase, generating a premature stop codon at positions
10–12 of this exon (Fig. 2). The owl monkey activating KIR
KIR Pseudogene KIR Inhibitory Model KIR Activating Model FcAR Model Exons LTR LINE SINE DNA Transposon
3DP1
3DL1
3DS1 3DS2
3DS3
3DL2
C -
- T
3DP1 3DL1 3DS1 3DL2 3DL3 4DS1 4DL1 4DL2
C -
- T - T
Ateles geoffroyi KIR haplotype
Aotus sp. KIR haplotype
Fig. 1 Gene organization of the KIR cluster in the spider monkey
(Ateles geoffroyi) and the owl monkey (Aotus sp.) based on BAC clone
draft sequences AC234015 and AC234076/AC193069, respectively.
Transcription orientation is indicated for each gene model, as well as
exon, repetitive element, and retroelement localization along the chro-
mosomal region. The predicted receptor structure is shown above each
gene model
Fig. 2 Ateles geoffroyi KIR gene models predicted to encode activating
receptors have a unique mechanism to generate truncated cytoplasmic
domains. The mechanism involves a single nucleotide deletion at posi-
tion 103 of exon 7 (box), inactivation of the donor splice site 1 (gray
background) of intron 7 by a G/C substitution at the first position of
intron 7 (gray box), a single nucleotide deletion at position 58 of intron
7(box), and the activation of a cryptic donor splice site 2 in intron 7 at
positions 66–73 (gray background). These alterations generated recep-
tors with cytoplasmic domains having a 22-amino acid insertion in the
cytoplasmic domain derived from intron 7 and a premature termination
due to a stop codon in exon 8
Immunogenetics

gene model Om-KIR4DS1 had the same modifications in
exon 7 and intron 7 (Fig. 2), suggesting that this mechanism
of switching from an inhibitory to an activating receptor is
prevalent in New World primates. Finally, the last KIR gene
model at the 3′ end of the cluster (Atge-KIR3DL2) was
predicted to encode a receptor with three Ig domains and a
long cytoplasmic tail with two putative ITIMs, suggesting
that it functions as an inhibitory receptor.
Atelidae express a variable set of activating and inhibitory
KIRs
A total of 29 unique full-length KIR cDNAs were sequenced
in the three Atelidae species, 13 corresponding to the two A.
hybridus individuals, 4 to the A. belzebuth individual, and 12
to the two L. lagotricha individuals (Supplementary Fig. 1).
These cDNAs were predicted to encode receptors with a
D0+D1+D2 configuration with long or short cytoplasmic
domains, with the only exception of a cDNA from A.
belzebuth that displayed a D1+D2 configuration with short
cytoplasmic domain. In addition, four types of alternatively
spliced variants (SV1-4) were identified in A. hybridus and
A. belzebuth (Supplementary Fig. 1). SV1 lacked exon 7
encoding the transmembrane domain, SV2 lacked exon 6
encoding the stem domain, SV3 lacked 36 bp at the 5′ end of
exon 4 (D1 domain), and SV4 lacked the last 200 bp of exon
3 (D0 domain). The structure of predicted KIR proteins was
modeled by homology, showing a highly similar folding to
that of KIR3DL1*001 (PDB 3VH8, average RMSD=0.076 Å,
Supplementary Fig. 2). The 13 full-length cDNAs from the A.
hybridus individuals were assigned to 5 putative loci based on
the phylogenetic analyses (see below) and distinctive sequence
features (Table 1). Three of these loci (Athy-KIR3DL1-3)were
predicted to encode inhibitory receptors having long cytoplasmic
domains and two ITIMs, while the other two loci (Athy-
KIR3DS1-2) were predicted to encode activating receptors with
short cytoplasmic domains with the intronic insertion at the
cytoplasmic domain describ ed above for the gene models Atge-
KIR3DS1-3. The transmembrane domain of these activating
receptors had the positively charged amino acid arginine at the
fourth position instead of a lysine at the ninth position, typical of
human activating KIRs (Lanier 1998). Athy-KIR3DL1
and Athy-
KIR3DS2 lacked exon 6 encoding the stem domain, a feature
shared by lineage V KIRs, represented by the huma n KIR3DL3
(Palacios et al. 201 1). The nucleotide similarity between the five
A. hybridus putative loci averaged 90.1 % (94.8–86.6 %, average
distance d=0.105), similar to that observed between A. geoffroyi
gene models (89 %, d=0.131). All five A. hybridus putative KIR
loci proved to be polymorphic in the three individuals analyz ed,
expressing two to four alleles per locus with an average
nucleotide similarity of 94 % (Table 1). In addition, the
three individuals analyzed had different haplotypes, with only
one individual (190) having cDNA sequences from the five
loci, whereas individuals 110 and 180 lacked sequences from
Athy-KIR3DS2 and Athy-KIR3DL1 loci, respectively (Table 1).
A. belzebuth cDNAs were assigned to three putative loci
(Table 1) predicted to encode an inhibitory receptor (Atbe-
KIR3DL2) and two activating receptors (Atbe-KIR3DS3 and
Atbe-KIR2DS3). Although Atbe-KIR2DS3 was the only
Atelidae KIR gene predicted to encode a receptor with a
D1+D2 configuration, it could represent a splicing variant
of Atbe-KIR3DS3 given their high sequence similarity (Sup-
plementary Fig. 1). In this species, the interlocus nucleotide
similarity averaged 95.1 % (d=0.051) and the mean allelic
similarity of Atbe-KIR3DL2, the only locus with more than
one allele, was 99.3 %. Finally, the 12 cDNAs sequences
from L. lagotricha were assigned to 6 putative loci (Table 1),
three of which were predicted to encode inhibitory receptors
(Lala-KIR3DL1
-3) and the other 3 where predicted to encode
activating receptors (Lala-KIR3DS1-3). The latter had
Table 1 Putative KIR loci/alleles assigned to the Atelidae cDNA sequences
Individual Putative KIR loci/alleles
b
A. hybridus
a
3DL1 3DS1 3DS2 3DL2 3DL3
110 01 02/03/sv1 01 sv4
180 02/04/sv1 01 sv3 01/02/sv4
190 02 01/sv1 02 02 03/sv4
A. belzebuth 3DS1 2DS1 3DL2
170 01 01/sv1/sv2 01/02/sv3
L. lagotricha 3DL1 3DS1 3DS2(?)
c
3DS3 3DL2 3DL3
300 01/03 02 01 02/04 01
660 01/02 01 01 01 01/03 01
a
Individual identification number for each species
b
Putative loci (italic type) were assigned according to clustering patterns in gene trees and sequence distinctive features. For each putative locus,
alleles or splicing variants (sv) were designated by numbers (01, 02, etc.)
c
Uncertainty about its status as a locus as it might be an allele of Lala-KIR3DL1
Immunogenetics
arginine at the fourth position of the transmembrane domain,
the intronic insertion at the cytoplasmic domain, and had in
common a unique single amino acid deletion at position 16th,
a feature shared by human inhibitory receptors KIR2DL1 and
Athy-KIR3DL2*02
Lala-KIR3DL1*03
Atbe-KIR3DL2sv3
Athy-KIR3DL2*01
Athy-KIR3DS1*03
Athy-KIR3DS1*04
Lala-KIR3DL3*01
Athy-KIR3DL3sv4
Athy-KIR3DS1*02
Lala-KIR3DS1*02
Atbe-KIR3DL2*01
Atbe-KIR2DS3*01
Lala-KIR3DL2*02
Lala-KIR3DL1*02
Lala-KIR3DL2*04
Athy-KIR3DL3*03
Athy-KIR3DL3*01
Lala-KIR3DS3*01
Lala-KIR3DL2*03
Lala-KIR3DS1*01
Athy-KIR3DS2*02
Lala-KIR3DS2*01
Atbe-KIR3DL2*02
Athy-KIR3DS1sv1
Atbe-KIR2DS3sv1
Atbe-KIR3DS3*01
Lala-KIR3DL2*01
Athy-KIR3DS2*01
Athy-KIR3DL3*02
Athy-KIR3DL2sv3
Lala-KIR3DL1*01
Atbe-KIR2DS3sv2
Athy-KIR3DS1*01
Om-KIR3DL2
Athy-KIR3DL1*02
Athy-KIR3DL1*01
Om-KIR4DS1
Om-KIR4DL1
Om-KIR3DL3
Om-KIR3DP1
Om-KIR4DL2
Om-KIR3DS1
Om-KIR3DL1
a
b
Immunogenetics

KIR2DL2. The interlocus nucleotide similarity between L.
lagotricha KIRs averaged 93.6 % (d=0.07) whereas the mean
allelic nucleotide similarity was 96.5 %. Lala-KIR3DS2 was
highly similar to Lala-KIR3DL1 , but it had a truncated cyto-
plasmic tail due to a C to T substitution in exon 9 that generated
a premature stop codon upstream from the region encoding the
proximal ITIM (Supplementary Fig. 1). This cDNA, however ,
did not encode for any positively char ged amino acid at the
transmembrane domain, and, based on sequence similarity, it is
possible that Lala-KIR3DL1 and Lala-KIR3DS2 segregate as
alleles of the same locus. Lala-KIR3DL2 cDNAs were unique
in that they had a 42-bp non-intronic insertion at the end of exon
5 that did not change the reading frame (Supplementary Fig. 1).
Diversity of KIR cytoplasmic domains in Atelidae
Predicted inhibitory receptors in Platyrrhini were grouped into
three classes according to the length and presence of regula-
tory motifs in their cytoplasmic domains (Supplementary
Fig. 3). The first class included sequences predicted to encode
receptors with cytoplasmic domains 83 residues long, identi-
cal in size to the human KIR3DL1, and included Athy-
KIR3DL2, Lala-KIR3DL2, and the 5 owl monkey KIR recep-
tors with long cytoplasmic domains. The second class were
characterized by a 5-bp deletion at the end exon 9 that shifted
the reading frame generating a cytoplasmic domain longer by
five residues than receptors from the first class (88 amino
acidslong).ThisclassincludedAtge-KIR3DL2, Athy-KIR3DL3,
Atbe-KIR3DL2, Lala-KIR3DL1,andLala-KIR3DL3 cDNAs.
Interestingly, one of the four alleles from the Lala-KIR3DL2
locus (Lala-KIR3DL2*04) encoded a cytoplasmic tail from the
second class, while the other three alleles were from the first
class, suggesting that either the deletion segregates in a single
locus or that the cDNA is a recombinant sequence (Suppleme n-
tary Fig. 1). Although cDNA sequences from class 2 did not
reach the stop codon, the genomic gene model from A. geoffr oyi
Atge-KIR3DL2 had this class of cytoplasmic domain and showed
the stop codon. The third class of Platyrrhini KIR3DL sequences
had a 4-b p deletion at the end of exon 9 , located 7 bp upstream
from the deletion seen in the second class of KIR3DL sequences.
This deletion shifted the reading frame generating predicted
cytoplasmic domains 114 amino acids long. Sequences from this
class included Atge-KIR3DL1 and Athy-KIR3DL1. Interestingly,
KIR genes from line age I (KIR2DL4/KIR2DL5)alsohada4-bp
deletion at a homologous site, coding for receptors with longer
cytoplasmic domains than those from other lineages (Campbell
and Purdy 201 1). The three classes of long cytoplasmic domains
shared some predicted function al motifs, including three protein
kinase C (PKC) phosphorylation sites (positions 4–6, 46–48, and
54–46), a N-myris toylation site (17–22), and a cAMP-/cGMP-
Table 2 Test of positive selection in Platyrrhini KIR genes
Taxon Test of selection
a
Sites under positive selection
b
n lnL M1 lnL M2 −2lnΔL p value
A. hybridus 13 −2,911.34 −2,904.29 14.1 8.7e−4 16, 32, 140, 147, 180, 226, 239, 241, 276, 277, 278
A. belzebuth 4 −1,786.19 −1,786.19 0 1 None
A. geofroyii 5 −2,594.66 −2,591.37 6.58 3.7e−3 32, 180, 199, 201, 258, 277
L. lagotricha 12 −2,728.41 −2,828.39 0.04 0.98 199, 240, 268
Aotus sp. 7 −2,938.59 −2,938.1 0.98 0.98 19, 20, 32, 110, 140, 143, 167, 215
Platyrrhini Clade 1 14 −2,784.58 −2,776.24 16.68 2.4e−4 16, 19, 27, 45, 47, 80, 164, 180, 237, 239, 240, 277, 278
Platyrrhini Clade 2 17 −2,965.82 −2,964.8 2.04 0.36 147, 180, 199, 226, 240, 277
Platyrrhini Clade 3 5 −2,394.39 −2,393.15 2.48 0.29 20, 143
Platyrrhini Cade 4 5 −2,214.02 −2,213.96 0.12 0.94 164,
167, 241
All Platyrrhini 41 −6,699.92 −6,685.81 28.22 7.4e−7 16, 19, 20, 22, 27, 32, 49, 52, 53, 54, 76, 91, 93, 95, 109,
110, 129, 140, 143, 145, 147, 165, 166, 167, 169, 180,
197, 199, 214, 215, 226, 236, 237, 240, 241, 252, 258,
260, 268, 276, 277, 278, 282
a
Likelihood ratio test of null hypothesis of neutrality (M1) versus alternative hypothesis of positive selection (M2)
b
Only sites from D0, D1, and D2 domains detected at the 0.1 significance level. Sites in italics are positions predicted to interact with MHC class I
molecules
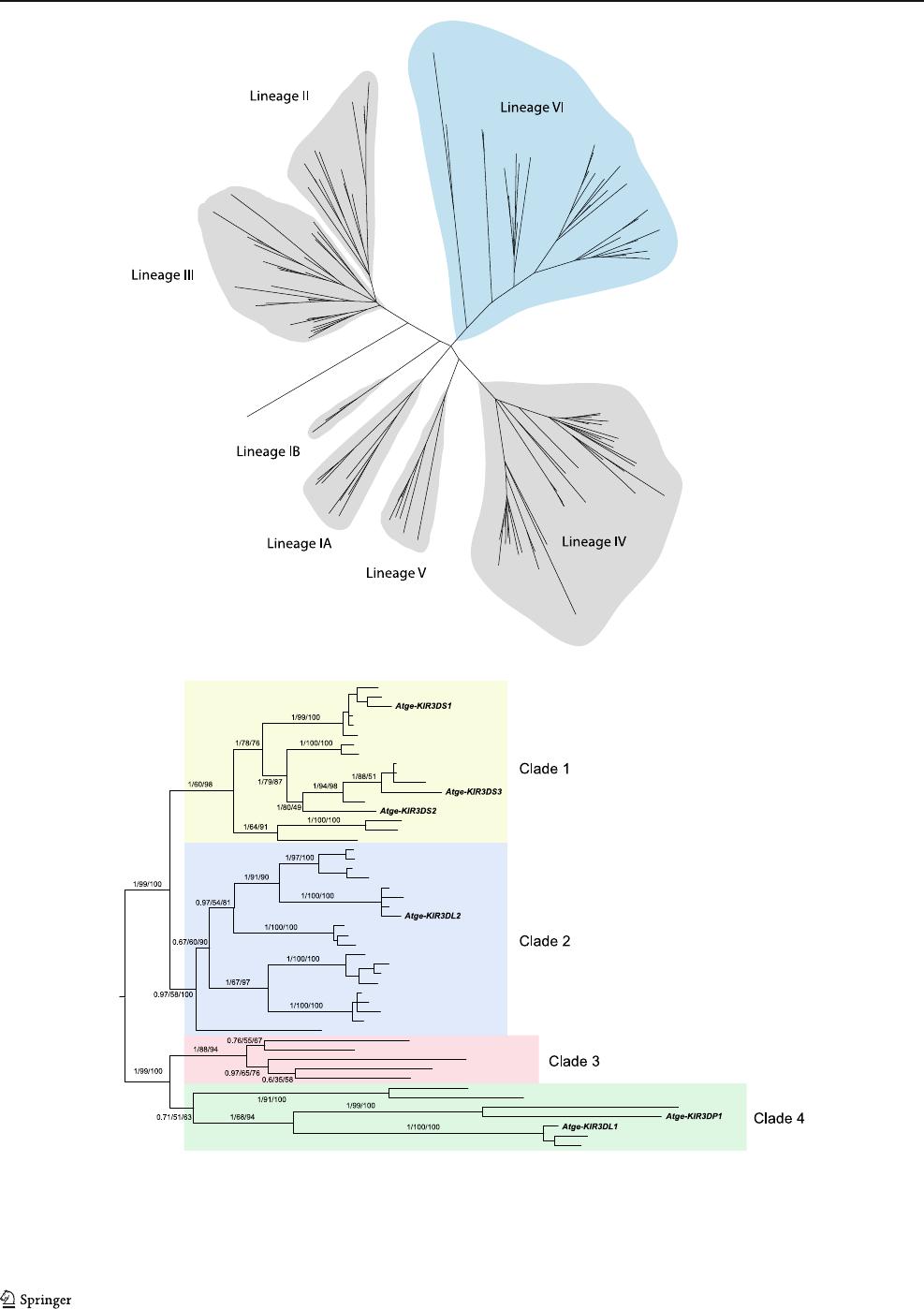
Fig. 3 Bayesian phylogenetic trees of primate KIR cDNAs and gene
models. a Platyrrhini KIRs form a monophyletic group conforming the
evolutionary lineage VI (blue background), with the Catarrhini evolu-
tionary lineages ( gray background) organized as sister groups. Lineage
IV, containing Old World primate KIRs, is considered a lineage II
subgroup based on intronic sequences (Guethlein et al. 2007b; Blokhuis
et al. 2011). b KIR cDNAs and gene models from Atelidae species and
from the owl monkey are clustered into four main clades. A. geoffroyi
(Atge) KIRs are shown in black type, A. hybridus (Athy) in red, A.
belzebuth (Atbe)inblue, L. lagotricha (Lala)ingreen, and owl monkey
(Om)inpurple. Numbers of branches indicate posterior probability for
Bayesian topologies, and bootstrap percentages after 1,000 replications
for maximum likelihood and neighbor-joining topologies, respectively
R
Immunogenetics
dependent protein kinase phosphoryla tion site (Supplementa ry
Fig. 3). They, however, differed in some functional motifs
including a unique PKC phosphorylation site (position 18–20)
in class 1 cytoplasmic domains, a unique casein kinase II
a
b
Immunogenetics

phosphorylation site (83–86) in class 2 cytopla smic domains,
and a PKC (92–94) and Casein kinase II (92–95) phosphoryla-
tion sites, an additiona l N-myristoylation site (109–1 14), and an
N-glycosylation site (108–1 1 1) in class 3 cytoplasmic domains.
The variation in regulatory sites between the classes of cytoplas-
mic domains suggests that inhibitory KIR receptors might have
alternative signaling routes conducing to differe nt functional
behaviors.
In contrast to inhibitory KIR receptors, all predicted acti-
vating KIRs in the Atelidae species had the same type of
truncated cytoplasmic domains. Their cDNAs had a single
nucleotide deletion three positions before the end of exon 7
and a 65-bp intronic insertion at the end of exon 7, generat-
ing a shift in the reading frame and a premature stop codon in
exon 8 (Supplementary Fig. 1), just as it was described for
the Atge-KIR3DS gene models (Fig. 2). A. hybridus and A.
belzebuth KIR3DS loci had a splice variant (SV1) that
skipped exon 7 and mai ntained the reading fram e to generate
predicted proteins with no transmembrane domain but with
long cytoplasmic domain having two ITIMs (Supplementary
Fig. 1). This supports the notion that modifications in exon 7
and intron 7 were responsible to generate truncated cytoplas-
mic domains in Atelidae activating KIRs. The only excep-
tion to this type of short tail KIRs in Atelidae was Lala-
KIR3DS2*01, which had a C/T nonsense mutation at the
region coding the proximal ITIM (Supplementary Fig. 1 ).
However, it lacked a positively charged residue in the trans-
membrane domain, questioning its role as an activating
receptor and suggesting that it is probably an allele from
the Lala-KIR3DL1 locus.
Platyrrhini KIRs form an independent evolutionary lineage
and are organized into four clades
Gene trees generated by distance, maximum likelihood, and
Bayesian approaches showed that Platyrrhini KIRs formed a
monophyletic group with no direct orthology to any of their
Catarrhini counterparts (Fig. 3a). This Platyrrhini clade
conformed the primate KIR lineage VI previously reported
(Cadavid and Lun 2009 ), which likely diversified from the
ancestral primate KIRs soon after the divergence between
Catarrhini and Platyrrhini some 44 million year ago.
Platyrrhini sequences conforming the KIR lineage VI were
in turn organized into four main clades (Fig. 3b). Clade 1
included all KIR sequences predicted to encode activating
receptors from the four Atelidae species analyzed. Clade 2
contained sequences predicted to encode inhibitory receptors
from the four Atelidae species, with the single exception of
the L. lagotricha cDNA Lala-KIR3DS2*01
, which, as men-
tioned be fore, might be an allele of the putat ive inhibitory
locus Lala-KIR3DL1. Clade 3 contained all the telomeric
gene models from the owl monkey and clade 4 clustered the
inverted centromeric gene models from the owl monkey
(Om-KIR3DP1, Om-KIR3DS1, and Om-KIR3DL2) and from
A. geoffroyi (Atge-KIR3DL1 and Atge-KIR3DP1), together
with the two A. hybridus Athy-KIR3DL1 alleles. Thus, clade 4
was the only containing sequences from Atelidae and the owl
monkey , suggesting that the most ancestral Platyrrhini KIRs are
likely to be those located in the inverted centromeric region of the
locus. However, no representative sequences from A. belzebuth or
L. Lagotricha were found in clade 4, suggesting either an incom-
plete sampling in these two species or the absence of the centro-
meric loci in those particular haplotypes. At least five orthologous
groups were identified from the trees, Athy-KIR3DS1/Atge-
KIR3DS1, Atbe-KIR3DS3/Atge-KIR3DS3, Atbe-KIR3DL2/Atge-
KIR3DL2/Athy-KIR3DL3, Athy-KIR3DL1/Atge-KIR3DL1,and
Atge-KIR3DP1/Aona-KIR3DP1 (Fig. 3b). No direct orthologous
genes were identified between L. lagotricha and the other Atelidae
KIRs, suggesting that the KIR locus in this family has diversified
very rapidly in a genus-specific fashion.
Rapid diversification of KIR genes in Atelidae
In order to understand the evolutionary forces that might
explain the rapid diversification of Platyrrhini KIRs,two
complementary approaches were used to detect signatures
of natural selection operating on this gene family. The first
method (PARRIS) tested the hypothesis of dN/dS>1 for the
entire sequence, while the other met hod (MEME) evaluated
both episodic and pervasive selection at the level of individ-
ual codon positions. Alignment-wise analysis of selection
was done by species, phylogenetic clades, and with the entire
Platyrrhini dataset (Table 2). Evidence of positive selection
was found at significant levels only in comparisons from A.
hybridus, A. geoffroyi, clade 1, and with all Platyrrhini se-
quences. In addition, in all but one category (A. belzebuth
sequences), there were individual sites under positive selec-
tion, including positions predicted to interact with the MHC
class I molecules (Table 2). Forty-three positively selected
sites were detected with the complete Platyrrhini dataset at
the 0.1 significance level, with 33 of them at the 0.05 signif-
icance level. These positions with higher significance were
evenly distributed between D0 and D1 domains, with 10 and
9 positions, respectively, but were more frequent in the D2
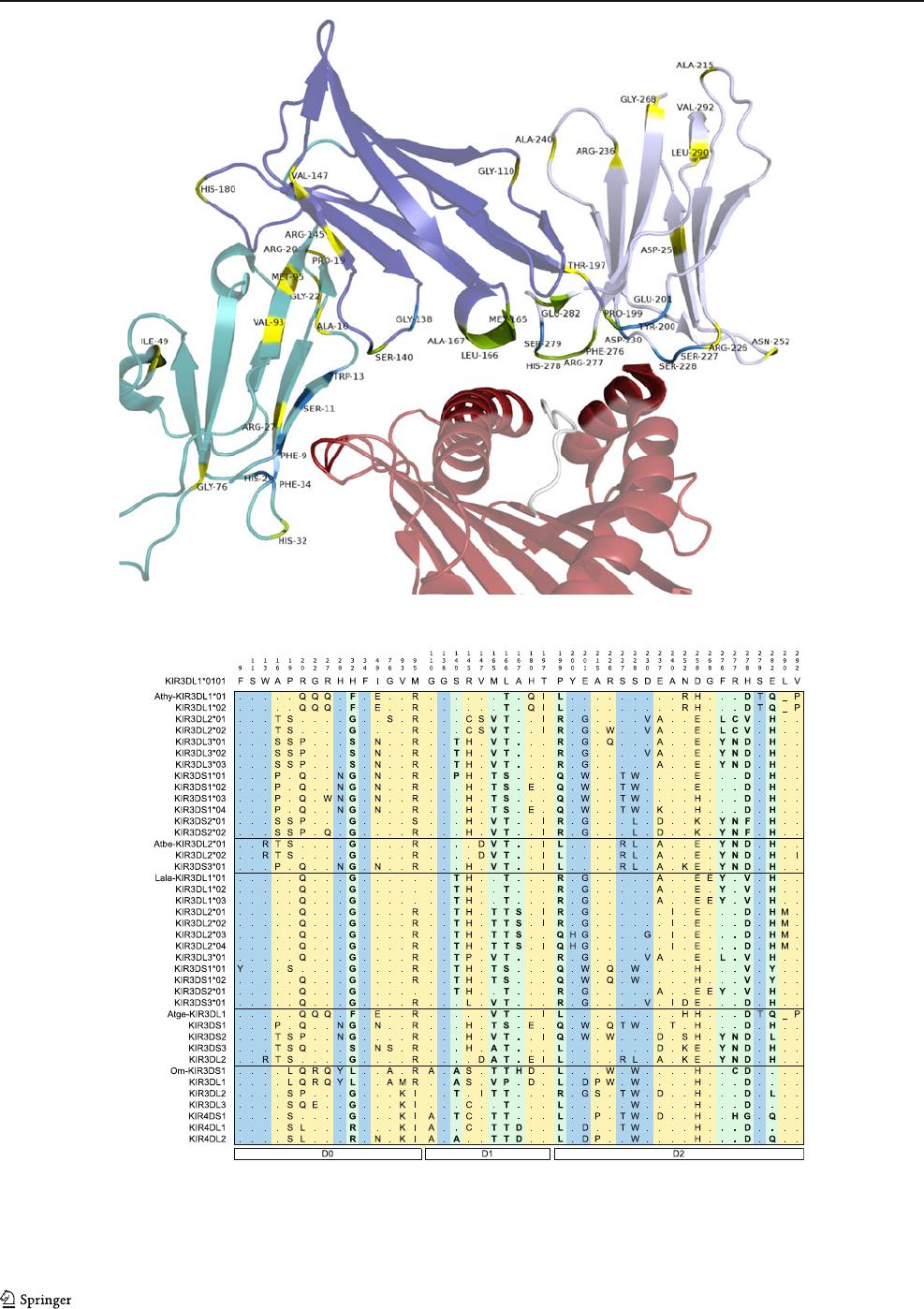
Fig. 4 Diversification of KIR loci in Platyrrhini has been favored by
positive selection. a Positively selected positions in D0, D1, and D2
domains identified in comparisons between all Platyrrhini sequences
at a significance level of p<0.05 are indicated over the human
KIR3DL1*0101-HLA-B27*01 structure (PDB 3VH8). Positions un-
der positive selection that are predicted to contact the MHC are
highlighted in green, whereas those that are not in contact with the
MHC are shown in yellow. Other positions that contact the MHC but
are not positively selected are highlighted in blue. The D0 domain is
shown in green,D1inblue,D2inlight purple, the MHC’s peptide
binding region in red, and the peptide in white. b Variation in
positions under positive selection in all Platyrrhini KIR sequences.
Positions are color-coded as in a, with positions under positive
selection that contact the pMHC in bold type
R
Immunogenetics

domain, with 14 positions (Fig. 4a). Ten of these positively
selected positions were predicted to interact with the MHC-
peptide (pMHC) complex, including a position interacting
with the beta-2 microglo bulin (residue 32) and positions
within the D1–D2 domains that conformed the three loops
of high variability predicted to affect the receptor specificity
for the pMHC (positions 165–167, 199 – 201, and 278–282)
(Vivian et al. 2011). In addition, four other positively select-
ed positions (16, 197, 226, and 252) were likely to affect
binding, although they were not in direct contact with the
pMHC complex (Fig. 4a ). The majority of the positions
subjected to positive selection (26 out of the 33), including
those contacting the pMHC, was variable between loci from
the same species but fixed between alleles of the same locus
(for example, positions 16 and 20 in A. hybridus loci, Fig. 4b).
On the contrary, only eight of the positively selected positions
were variable between alleles of the same locus in at least one
species (for example, position 199 in Lala-KIR3DL2 alleles,
Fig. 4b). In addition, 7 out of the 10 positively selected
positions that were part of the interacting interface with the
pMHC (32, 140, 167, 276, 277, 278, and 282) were fixed in at
least one species, indicating species specificity in the KIR
mode of evolution. Finally, there was a general tendency among
putative orthologous groups (i.e., Athy-KIR3DL1/Atge-KIR3DL1
Athy-KIR3DS1/Atge-KIR3DS1) to share the same residues at the
pMHC-contacting interface, indicating that they might be binding
the same type of MHC class I ligands. T ogether , these data indicate
that diversifying selection might be more important to generate
differences in binding affinity and specificity between receptors of
the KIR cluster than to alleles of the same locus.
Discussion
The analysis of the KIR family in four Atelidae species
showed that the system is constituted by a variable set of
activating and inhibitory receptors expanded from the ances-
tral primate lineage but that diversified independently from
their Catarrhini counterparts. The KIR locus in A. geoffroyi
contained at least six KIR gene models with a D0+D1+D2
configuration having long or short cytoplasmic domains.
The two most centromeric gene models (Atge-KIR3DP1
and Atge-KIR3DL1) had an opposite transcription orienta-
tion, similar to what was observ ed in the KIR family of the
owl monkey, and thus, it constitutes a unique feature of New
World primate KIR haplotypes studied to date. The centro-
meric gene models from A. geoffroyi and the owl monkey
formed a monophyletic group, indicating that they are the
most ancestral Platyrrhini KIRs and that the inversion of the
centromeric region occurred before the divergence of
Atelidae and Aotinae taxa at least 20 MYA. The fact that
no orthologous genes from the centromeric loci were found
in A. belzebuth or L. lagotricha individuals might be
explained by either an insufficient plasmid clone sampling
or an actual absence of those genes in those species.
The other four gene models of the A. geoffroyi KIR hap-
lotype (Atge
-3DS1-3 and Atge-KIR3DL2) had the typical
transcription orientation of primate KIRs and were not
orthologous to any of the owl monkey gene models. The
three predicted activating receptors were generated by a
unique mechanism involving a single nucleotide deletion at
the end of exon 7, the eliminatio n of the donor splicing site of
intron 7, and the activation of a secondary donor splicing site
in the same intron, together generating a frame shift, the
incorporation of an intronic segment, and a premature stop
codon in exon 8. All other activating KIRs in Atelidae were
also generated by this mechanism, and they clustered togeth-
er in gene trees, suggesting that activating KIRs in Atelidae
originated once from an ancestral inhibitory KIR and subse-
quently diversified in a genus-specific fashion by gene du-
plication events. Indeed, one of the two owl monkey KIR
gene models predicted to encode activating KIRs (Om-
KIR4DS1) was generated by this mechanism, but did not
cluster with the Atelidae-activating KIRs, supporting a trend
of rapid genus-specific diversification. Activating KIRs in
primates, therefore, have evolved recurrently and through
different mechanisms from inhibitory receptors (Abi-Rached
and Parham 2005).
Atelidae telomeric KIR3DL sequences formed a monophy-
letic clade with the KIR3DS genes as a sister group, but L.
lagotricha genes cluster independently form the other
Atelidae inhibitory receptors, indicating the lack of orthology
of KIR genes between genera and further supporting a mode
of evolution characterized by a rapid genus-specific diversifi-
cation of the KIR locus telomeric region. This diversification
is explained by an active process of gene duplication and
divergence, likely promoted by selection for ligand binding.
Indeed, signatures of positive selection in paralogous genes
were detected in a large fraction of residues predicted to
interact with variable residues of the MHC class I molecules
based on the human KIR-MHC structure. Such signatures,
however, were not detected between alleles of the same locus,
indicating that positive selection has acted primarily to pro-
mote divergence between loci but not to generate polymor-
phism. Thus, the major evolutionary force promoting the
diversification of Platyrrhini KIR loci appear to be the affinity
interaction with the highly variable set of G- and B-like MHC
class I molecules known to be present in this taxon (Sawai
et al. 2004).
The KIR family has evolved by a dynamic process of gene
expansion and contraction (Hao and Nei 2005). In mammals,
at least two ancestral lineages originated the extant KIRs.
One is represented by the KIR3DX1 locus located in the
central part of the LILR gene family (Sambrook et al.
2006), which is single copied in humans but has been ex-
panded in cattle (Dobromylskyj and Ellis 2007). The other
Immunogenetics
