
Phase Transformation of Calcium Phosphates
by Electrodeposition and Heat Treatment
WEI-JEN SHIH, MOO-CHIN WANG, KUO-MING CHANG, CHENG-LI WANG,
SZU-HAO WANG, WANG-LONG LI, and HONG-HSIN HUANG
The effect of heat treatment on the calcium phosphate deposited on Ti-6Al-4V substrate using
an electrolytic process is investigated. The calcium phosphate was deposited in a 0.04 M
Ca(H
2
PO
4
)
2
ÆH
2
O (MCPM) solution on a Ti-6Al-4V substrate at 333 K (60 C), 10 V, and 80
Torr for 1 hour, and calcined at various temperatures for 4 hours. The X-ray diffraction (XRD)
results demonstrate that the phases are dicalcium phosphate (CaHPO
4
, DCPD) and hydrox-
yapatile [Ca(PO
4
)
6
(OH)
2
, HAP] for the as-deposited samples. When the deposited sample was
calcined at 873 K (600 C) for 4 hours, the XRD results show that the transformation of DCPD
to HAP occurs. Moreover, HAP converts to b-TCP, CPP, and CaO. For the sample calcined at
1073 K (800 C) for 4 hours, the scanning electron microscopy (SEM) micrograph reveals that
the crack of the calcined sample propagates with a width of about 3 lm. This result is due to
HAP becoming decomposed and converting to b-TCP, CPP, CaO, and H
2
O. The vaporization
of H
2
O within the calcined sample promotes the crack propagation and growth.
DOI: 10.1007/s11661-010-0417-x
The Minerals, Metals & Materials Society and ASM Internati onal 2010
I. INTRODUCTION
HYDROXYAPATITE ceramic (Ca
10
(PO
4
)
6
(OH)
2
,
hereafter HAP) is used in orthopaedics and dental
implant surgery, either alone or in combination with
other materials or substrates, as a coating on metal
implants
[1,2]
and to fill bone defects.
[2–4]
A lot of effort
has been made in recent years to develop processing
methods, such as plasma spraying,
[5,6]
electrophoretic
methods,
[7,8]
and electrochemical method s,
[9–12]
for
depositing calcium phosphate ceramics on the implant
substrate alloys in order to have high strength, good
processability, suitable specific density, and excellent
corrosion resistance in the living body.
Although the most widely applied HAP coating
procedure is the plasma spray technique,
[5,6]
the major
problem of the decomposition and phase transformation
of HAP during the spray coating process still exists.
Hence, the electrochemical deposition of calcium phos-
phate bioceramic coatings has attracted considerable
attention
[9–12]
because of its many advantages. Specifi-
cally, composi tion and coatin g structure controls are
possible due to the relatively low processing temperature,
and highly irregular objects can be coated relatively
quickly.
[13]
Since electrochemical deposition can be the
result of increasing pH at the interface, which is attrib-
uted to electron incorporation to form OH
–
ions and H
2
through water reduction, H
2
gas evolution at the interface
leads to a heterogeneous coati ng.
[13]
Recent work has
used organic solutions to avoid the negative effects of H
2
gas, and more homogeneous coatings have been reported
by Chen et al.
[14]
In addition, Wang et al.
[11]
pointed out
that when calcium phosphate coatings are deposited on a
Ti-6Al-4V substrate using an electrolytic method under
80 Torr, bubbles quickly lift from the cathode surface,
making the deposit regular and integrated.
When the electrolyte contains Ca
2+
and H
2
PO
4
1–
,it
produces calcium phosphate powder s, such as mono-
calcium phosphate monohydrate (MCPM, Ca(H
2
PO
4
)
2
Æ
H
2
O), dicalcium phosphate dihydrate (DCPD, CaHPO
4
Æ
2H
2
O), octacalcium phosphate, amorphous calcium
phosphate, and hydroxyapatite (HAP), depending on
the Ca/P ratio of raw materials and the reaction that
occurs.
[15,16]
Among these, HAP is the most interesting
form of calcium phosphate, and has been electrochem-
ically deposited from several solutions by a number of
researchers.
[9,11,17–20]
However, the effect of heat treat-
ment on HAP formation on a Ti-6Al-4V substrate in a
0.04 M MCPM solution using an electrolytic process
has not yet been discussed in detail.
In the present study, a 0.04 M MCPM solution was
used for the synthesis of HAP on a Ti-6Al-4V substrate
using an electrolytic process. The main objective of this
investigation is to study the effect of heat treatment on
HAP formation on a Ti-6Al-4V substrate by differential
thermal and thermogravimetric analyses (DTA/TGA),
Fourier transform infrared spectroscopy (FT-IR), X-ray
diffraction (XRD), scanning electron microscopy
WEI-JEN SHIH, Engineer, is with the Metal Industries Research
and Development Center, Kaohsiung 81160, Taiwan R.O.C.
MOO-CHIN WANG, Professor, is with the Head of Department of
Fragrance and Cosmetic Science, Kaohsiung Medical University,
Kaohsiung 80782, Taiwan R.O.C. Contact e-mail: mcwang@kmu.
edu.tw KUO-MING CHANG, Professor, CHENG-LI WANG, PhD
Student, and SZU-HAO WANG, Engineer, are with the Department
of Mechanical Engineering, National Kaohsiung University of
Applied Sciences, Kaohsiung 80782, Taiwan R.O.C. WANG-LONG
LI, Professor, is with the Institute of Nanotechnology and Micro-
systems Engineering, National Cheng Kung University, Tainan 70101,
Taiwan R.O.C. HONG-HSIN HUANG, Professor, is with the
Department of Electrical Engineering, Cheng Shiu University,
Niaosong, Kaohsiung 83347, Taiwan R.O.C.
Manuscript submitted July 8, 2009.
Article published online October 5, 2010
METALLURGICAL AND MATERIALS TRANSACTIONS A VOLUME 41A, DECEMBER 2010—3509
(SEM), transmission electron microscopy (TEM), and
electron diffraction (ED).
The purposes of this study are to (1) determine the
thermal behavior of the converted calcium phosphate
that is deposited, (2) investigate the phase transforma-
tion of the as-deposited and postcalcined calcium
phosphate samples, and (3) observe the microstructure
of the calcium phosphate deposited and postcalcined at
various temperatures.
II. EXPERIMENTAL PROCEDURE
A. Substrate Preparation
A Ti-6Al-4V alloy plate (ASTM standard F-136) and
a platinum plate were used as the cathode and anode,
respectively. A 15 9 15 9 3 mm Ti-6Al-4V alloy plate
was mechanically ground with SiC papers from 120 to
1200 grit and polished with 0.3-lmAl
2
O
3
powders to a
mirror finish. The Ti-6Al-4V plate was then washed
thoroughly with running distilled water before being
ultrasonically degreased with acetone and dried at
333 K (60 C).
B. Electrolytic Deposition
The saturated 0.04 M electrolyte was prepared by
adding 1 g of analytical grade monocalcium phosphate
monohydrate (Ca(H
2
PO
4
)
2
ÆH
2
O, MCPM; supplied by
Showa Chemical Co. Ltd., Tokyo) into 100 mL of
water. The electrolyte was stirred with a magnetic stirrer
for 1 hour to enhance the dissolution of the calcium
phosphate. The pH of the electrolyte was about 3.0.
Electrolytic deposition was carried out at 333 K (60 C)
for 20 to 120 minutes under a cathode voltage of 4 to
10 V. The distance between the electrodes and the
cathode area was maintained at 3 cm and 1.057 cm
2
,
respectively. The ambient pressure of 80 Torr was
selected for the electrolysis to improve the assembly of
the experimental setup, which is shown in Figure 1.
After deposition, the sample was was hed in distilled
water and dried in air at room temperature.
C. Sample Characterization
DTA/TGA was conducted on a 5.0-mg powder sam-
ple at a heating rate of 10 C/min in air (Simultaneous
symmetrical thermoanalyzer, TGA24, SETARAM,
Caluire, France) with Al
2
O
3
powders as a reference
material.
The chemical behavior and molecular bonding struc-
ture of the converted HAP were evaluated using a Fourier
transform infrared spectroscope (PerkinElmer Spectrum
One FT-IR spectrometer, Boston, MA). Each sample was
mixed with KBr (sample: KBr = 1 : 99 in mass ratio) and
was pressed into 200-mg pellets, 13 mm in diameter, for
taking infrared adsorption spectra at a frequency range of
400 to 4000 c m
1
. A spectral resolution of 4 cm
1
was
chosen, and the composite spectrum for each sample was
represented by the average of 64 scans, normalized to the
spectrum of the blank KBr pellets.
The crystalline phases of the dried and postcalcined
samples were examined using XRD (Rigaku D-Max/
IIIV, Tokyo). Monochromatic Cu K
a
radiation and a Ni
filter were selected. The operating tube voltage and
current were 30 kV and 20 mA, respectively. The
scanning angle (2h) of the sample was from 20 to
55 deg, with a scanning speed of 4 deg/min.
The coating microstructure and morphology were
investigated using a scanning electron microscope
(Hitachi S2700 SEM, Hitachi, Tokyo). A transmission
electron microscope (Hitachi FE-2000) was used to
determine the crystal structure at 200 kV. The TEM
sample was prepared by dispersing the HAP powders in
an ultrasonic bath and then collecting them on a copper
grid.
III. RESULTS AND DISCUSSION
A. Thermal Behavior of the Converted Calcium
Phosphate Deposits
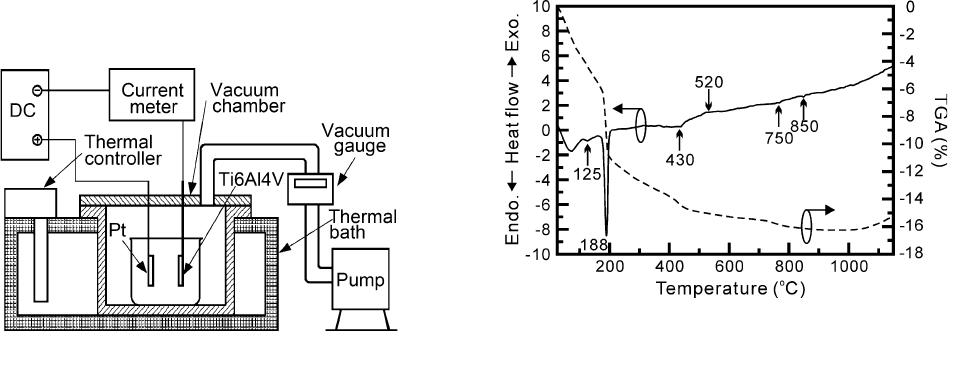
Figure 2 shows the DTA/TGA curves of the calcium
phosphate deposited under 10 V at 333 K (60 C) for
1 hour and measured at a heating rate of 10 C/min in
air. This figure indicates that the endothermic peaks at
398 K and 461 K (125 C and 188 C) accompanied by
weight losses of 3.0 and 4.8 pct, respectively, are
attributed to the vaporization of water. An endothermic
peak at around 703 K (430 C) accompanied by a
Fig. 2—DTA/TGA curves measured at a heating rate of 10 C/min
in air for calcium phosphate powders deposited at 333 K (60 C)
and 10 V for 1 h.
Fig. 1—Assembly diagram of low-pressure electrolytic deposition.
3510—VOLUME 41A, DECEMBER 2010 METALLURGICAL AND MATERIALS TRANSACTIONS A
weight loss of 1.2 pct (total weight loss of 14.8 pct) is
due to the release of crystal water of dicalcium phos-
phate dihydrate (DCPD, CaHPO
4
Æ2H
2
O).
[19]
An exo-
thermic reaction peak at 793 K (520 C) is attributed to
the crystallization of HAP. The weak broad arc-form
continuum of the exothermic reaction between 1023 K
and 1123 K (750 C and 850 C) is due to the form ation
of calcium pyrophosphate (Ca
2
P
2
O
7
) and b-tricalcium
phosphate (Ca
3
(PO
4
)
2
, b-TCP).
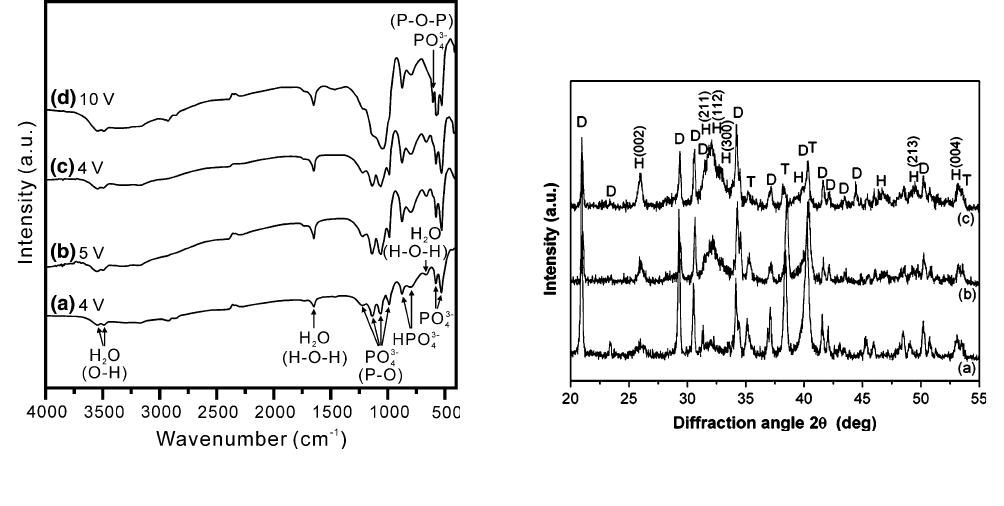
The FT-IR spectra for the calcium phosphate pow-
ders deposited at 333 K (60 C) under various applied
voltages for 1 hour are shown in Figure 3. For an
applied voltage of 4 V (Figure 3(a)), the absorptions at
3522 and 3488 cm
1
are attributed to absorbed water.
The characteristic bands around 1653 and 682 cm
1
are
consistent with H-O-H bonding vibration.
[19,20]
The
band at 1653 cm
1
is also due to water molecules and
the oxidized titanium layer on the metal.
[21]
The absorp-
tions at 1061, 1220, and 1135 cm
1
are due to P = O
associated stretching vibrations.
[19]
The P = O stretch-
ing vibration in PO
4
3–
ions at 987 cm
1
is observed. The
bands located in the range of 1090 to 1030 cm
1
and at
960 cm
1
are consistent with phosphate group absorp-
tion in HAP, as reported by Manso et al.
[20]
The bands
at 875 and 799 cm
1
are due to the P-O-P asymmetric
stretching vibration in the HPO
4
2–
group.
[19,21]
The
bands located at 578 and 527 cm
1
can be assigned to
the P-O mode of the PO
4
3–
characteristic peak. How-
ever, in the bands at 500 to 700 cm
1
, the most intense
peak observed at 600 cm
1
is not found in DCPD, but it
appears in all amorphous calcium phosphates, including
amorphous dicalcium phosphate.
[22]
In the present
study, the bands observed in this range could be
associated with the DC PD. When the applied voltage
is increased from 5 to 10 V (Figures 3(b) through (e)),
the band at 603 cm
1
represents the O-P-O bonding
vibrations of the PO
4
group in the phosphate
deposits.
[23]
The intensity of the 578 and 527 cm
1
peaks increases with applied voltage. This is because in
well-crystallized DCPD, the DCPD phosphate peaks
become progressively more clearly defined and intense,
and a spectrum analogous to that of well-crystallized
DCPD is eventually obtained.
[22]
B. Phase Transformation of the As-Deposited
and Postcalcined Calcium Phosphate Samples
XRD patterns of the calcium phosphate samples
deposited at 333 K (60 C) under 10 V for various
durations are shown in Figure 4. With a 20-minute
deposition time (Figure 4(a)), DCPD a nd the Ti (sub-
strate) are the dominant phases and the HAP is the
minor phase.
When the deposition time is increased from 20 to 60
and 120 minutes (Figures 4(b) and (c)), the phase does
not change, but the reflection intensity of HAP increases
with the deposition time. Figure 4 also indica tes that the
reflection intensity of Ti decreases with increasing
deposition time. This is because the thickness of the
deposits increases along with the deposition time, which
reduces Ti reflections at 2h = 37.8 and 39.6 deg.
The formation mechanism of DCPD and HAP can be
explained as follows. MCPM is the most soluble and
acidic among the calcium phosphates. The dissolution of
MCPM at temperatures from 298 K to 373 K (25 Cto
100 C) has been expressed as the followin g reaction
equation:
[24]
Ca H
2
PO
4
ðÞ
2
H
2
O þ xH
2
O
!
H
2
O
CaHPO
4
þ H
3
PO
4
þ 1 þ xðÞH
2
O ½1
The stepwise dissociation of H
3
PO
4
acid
[25]
is as
follows:
H
3
PO
4
! H
þ
þ H
2
PO
4
K
I
¼ 7:5 10
3
½2
H
2
PO
4
! H
þ
þ H
2
PO
4
K
II
¼ 6:2 10
8
½3
Fig. 3—FT-IR spectra for the calcium phosphate powders deposited
at 333 K (60 C) for 1 h under various applied voltages.
Fig. 4—XRD patterns of the calcium phosphate samples deposited
at 333 K (60 C) under 10 V for various durations: (a) 20 min,
(b) 60 min, and (c) 120 min (D: DCPD, H: HAP, and T: Ti).
METALLURGICAL AND MATERIALS TRANSACTIONS A VOLUME 41A, DECEMBER 2010—3511
HPO
2
4
! H
þ
þ PO
3
4
K
III
¼ 1:0 10
12
½4
where K
I
,K
II
, and K
III
denote the ionization constants
of H
3
PO
4
,H
2
PO
4
–
, and HPO
4
2–
in water, respectively.
Since H
2
PO
4
–
,HPO
4,
2–
and PO
4
3–
ions exist at the
electrode, the other related cathode reactions are as
follows:
[26]
2H
2
O þ2e
! H
2
þ 2OH
½5
2H
2
PO þ 2e
! 2HPO
2
4
þ H
2
½6
2HPO
2
4
þ 2e
! 2PO
3
4
þ H
2
½7
HPO
2
4
þ OH
! PO
3
4
þ H
2
O ½8
DCPD is formed on the cathode surface as expressed
by the following reaction:
Ca
2þ
þ HPO
2
4
þ 2H
2
O ! CaHPO
4
H
2
O ½9
It has been established that the electrolytic deposition
results in an increasing local pH within the diff usion
layer due to elect ron incorporation to form OH
–
ions
and H
2
through water reduction.
[13]
When excess OH
–
(Reaction [5]) is produced, phosphate ions needed for
HAP increase
[26]
and the following reaction takes place
on the cathode surface and then HAP could be
deposited.
10Ca
2þ
þ 6PO
3
4
þ 2OH
! Ca
10
PO
4
ðÞ
6
OHðÞ
2
½10
Figure 5 shows the solubility product diagram of
aqueous solutions of calcium phosphate,
[27]
in which it is
seen that when the deposition time is short (less than
20 minutes), the pH value is low and the HPO
4
2–
ions
are stable. Thus, Eqs. [6] and [9] are the major reactions
that make DCPD the dominant phase. The pH value
increases when the deposition duration is greater than
20 minutes for the OH
–
ions around the metal/solution
interface, and Eqs. [7] and [10] become the dominant
reactions
[28]
making the major phase of HAP formation.
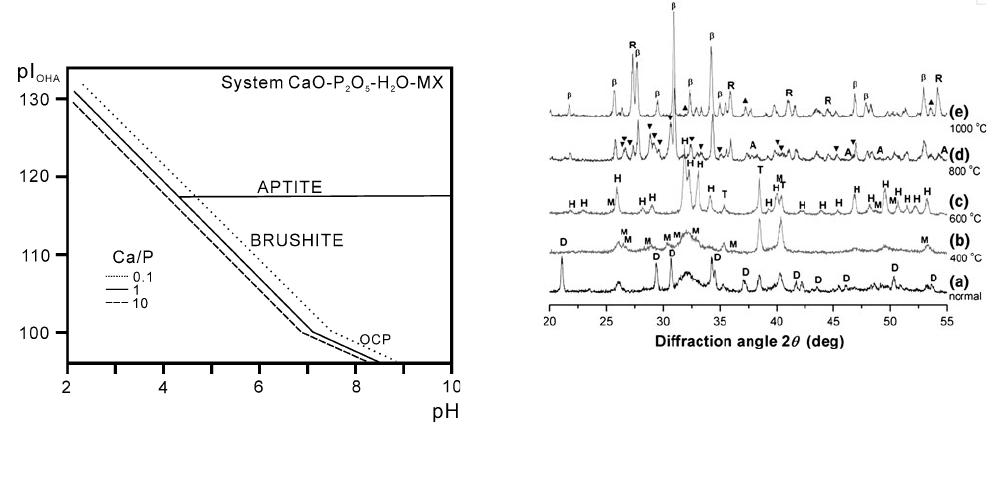
Figure 6 shows the XRD patterns of the calcium
phosphate samples deposited at 333 K (60 C) under
10 V for 1 hour and calcined at various temperatures for
4 hours. Figure 6(a) shows the XRD pattern of an as-
deposited sample that contains phases of DCPD and
HAP. The XRD pattern of the calcium phosphate
deposited sample calcined at 673 K (400 C) for 4 hours
is shown in Figure 6(b), where the DCPD phase
vanishes and HAP is the major phase.
Figure 6(c) shows the XRD pattern of the deposited
sample calcined at 873 K (600 C) for 4 hours, revealing
that HAP is the dominant phase, with some MCPM as
the minor phase. The reflection intensity of HAP (211),
(112), and (300) increases when the calcined temperatur e
rises from 673 K to 873 K (400 C to 600 C), and this
is because the intensity of the DCPD crystalline plane
disappears when it converts to a polycrystalline HAP.
In the DTA results, the exothermic react ion peak at
793 K (520 C) is attributed to the crystallization of
HAP. Hence, a more crystallized HAP compared with
as-deposited HAP can be obtained when the calcination
temperature is greater than 793 K (520 C). This result
is responsible for the well-defined peaks that indicate a
well-crystallized HAP.
Due to variations in the process parameters, such as
pH value, duration, and temperature, the amorphous
phosphate, or Ca
10
(HPO
4
)(PO
4
)
6
, is transformed into
defective hydroxyapatite (d-HAP, calcium-deficient
hydroxyapatite, Ca
10–x
(HPO
4
)
x
(PO
4
)
6–x
(OH)
2–x
0 £ x £ 1)
and stoichiometric HAP (s-HAP, Ca/P = 1.67).
[29]
The
relative quantities of the two products are determined by
the Ca/P ratio. If the ratio is near the theoretical ratio of
1.67 for HAP, the product may be s-HAP with a little
d-HAP. However, the lower the Ca/P ratio, the more
d-HAP will be found.
[30]
The d-HAP and s-HAP
materials have similar crystal structures and belong to
Fig. 5—Solubility product diagram of aqueous solutions of calcium
phosphates.
[27]
Fig. 6—XRD patterns of the calcium phosphate samples deposited
at 333 K (60 C) under 10 V for 1 h and calcined at various temper-
atures for 4 h: (a) as-deposited, (b) 673 K (400 C), (c) 873 K
(600 C), (d) 1073 K (800 C), and (e) 1273 K (1000 C) (D: DCPD,
H: HAP, C: CPP, T: Ti, A: anatase, R: rutile, b: b-TCP, O: CaO,
and M: MCPM).
3512—VOLUME 41A, DECEMBER 2010 METALLURGICAL AND MATERIALS TRANSACTIONS A
the same space group P6
3
/m. Their XRD patterns are
identical, and thus whether the product is pure HAP or
a mixture of d-HAP and s-HAP, the XRD patterns are
the same.
In the present study, the Ca/P ratio of the deposited
samples is 1.54, which is close to the theoretical Ca/P
ratio of 1.67 for HAP.
[11]
Hence, s-HAP is present in
large amounts and d-HAP in small amounts. The
transformation of DCPD to HAP at 873 K (600 C)
follows the reaction
20CaHPO
4
!
600
C
Ca
9
HPO
4
ðÞPO
4
ðÞ
5
OHðÞ
þ Ca
10
PO
4
ðÞ
6
OHðÞ
2
þCaO
þ 4P
2
O
5
þ 8H
2
O ½11
The XRD pattern of the deposited sample calcined at
1073 K (800 C) for 4 hours is shown in Figure 6(d). It
indicates that the crystallized phases are composed of
the major phases of b-TCP and CPP and a minor phase
of HAP. Moreover, the minor phases of CaO, anatase
(TiO
2
), and rutile (TiO
2
) also ap pear.
According to the DTA results, the e xothermic reac-
tion exists between 1023 K and 1123 K (750 Cand
850 C) and the associated weight loss is commensurate
with Eq. [11], which describes the conversion of HAP to
b-TCP. Although Kamiya et al.
[31]
reported that b-TCP
was obtained beyond 1073 K (800 C), HAP decom-
poses partly with the following reaction:
Ca
10
PO
4
ðÞ
6
OHðÞ
2
! 3Ca
3
PO
4
ðÞ
2
þCaO þ H
2
O ½12
However, Eq. [12] cannot show the CPP formation
and it cannot explain why only a part of HAP is
decomposed. In the present study, another decomposi-
tion of HAP must occur:
[32]
Ca
10
PO
4
ðÞ
6
OHðÞ
2
! 2Ca
3
PO
4
ðÞ
2
þCa
2
P
2
O
7
þ 2CaO
þ H
2
O ½13
Furthermore, d-HAP is decomposed as follows:
[30]
Ca
9
HPO
4
ðÞPO
4
ðÞ
5
OHðÞ!3Ca
3
PO
4
ðÞ
2
þH
2
O ½14
The XRD pattern of the deposited sample calcined at
1273 K (1000 C) for 4 hours is shown in Figure 6(e). It
is found that TiO
2
only has the rutile phase. Moreover,
the intensity of HAP and CPP decreases. b-TCP and
H
2
O are formed according to the following equation:
Ca
10
PO
4
ðÞ
6
OHðÞ
2
þCa
2
P
2
O
7
! 4Ca
3
PO
4
ðÞ
2
þH
2
O
½15
C. Microstructure of the Deposited Sample Calcined
at Various Temperatures for 4 Hours
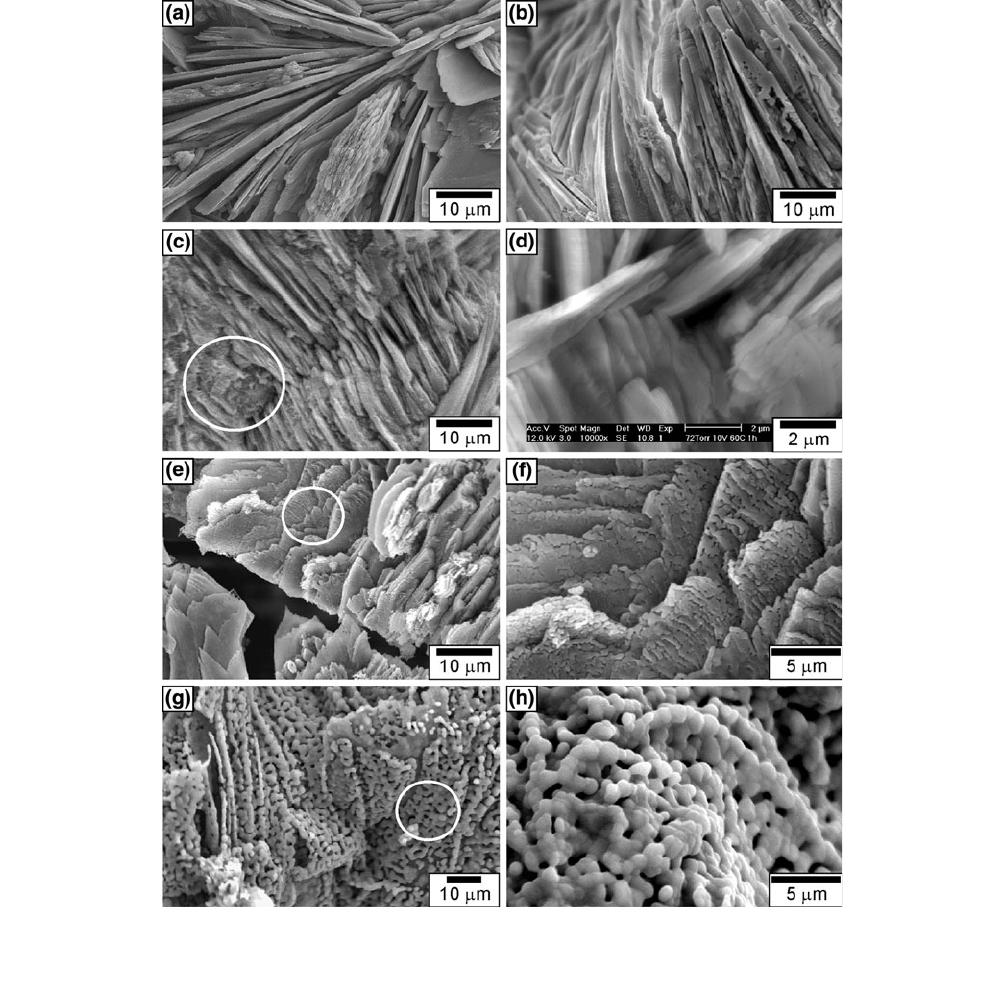
The SEM morphologies of the sample deposited at
10 V and 333 K (60 C) for 1 hour under 80 Torr and of
those calcined at various temperatures for 4 hours
are shown in Figure 7. Figure 7(a) shows that the
as-deposited sample of the needlelike DCPD crystals is
smooth, flat, and has a sharp edge, but parts of the tips
are round. Figure 7(b) shows the morphology of DCPD
after being calcined at 673 K (400 C) for 4 hours,
indicating an insignificant change in the morphology
compared with Figure 7(a). In Figure 7(b), the mor-
phology of DCPD is still needlelike, except for the 1-lm
microcrack on the surface. According to the results of
Figure 6(b), the crystal phase of DCPD still appears,
but without the crystal water. This phenomenon leads to
the shrinkage of the calcined sample and creat es the
microcrack.
The SEM morphology of the deposit ed samples
calcined at 873 K (600 C) for 4 hours is shown in
Figure 7(c). It is found that the HAP crystals forming
on the needlelike crystals have a significant change in
morphology. Figu re 7(d) shows an enlarged view of the
white circle in Figure 7(c). It is found that the platelike
crystals have a length and width of about 7.0 and
2.0 lm, respectively. Ban and Hasegawa
[12]
noted that
some of the deposits formed at 373 K (100 C) for
10 minutes were platelike, and others needlelike. XRD
results confirmed that all crystals, including platelike
ones, were HAP growing along the c-axis. This result
agrees with the results in Figure 6(c) and those reported
by Ban and Haseqawa.
[12]
The SEM micrograph of a sample calcined at 1073 K
(800 C) for 4 hours is shown in Figure 7(e). It reveal s
that the crack in the calcined sample propagates with a
width of about 3 lm. This result is due to HAP being
decomposed and converting to b-TCP, CPP, CaO, and
H
2
O. The vaporization of H
2
O within the calcined
sample promotes the crack propagation and growth.
However, the deposited sample calcined at 1073 K
(800 C) for 4 hours has a number of significant
problems, including phase/chemical decomposition of
HAP, absence of a chemical interface/bond between the
coating and substrate, thick coating, and cracks/lami-
nation through the coating.
[10]
Figure 7(f) shows the
enlarged view of Figure 7(e), revealing that the platelike
crystals convert into elongat e-type ones with pores. This
result corresponds to the CPP obtained for calcination
at higher temperatures.
[33]
Figure 7(g) shows the SEM morphology of the
deposited sample calcined at 1273 K (1000 C) for
4 hours. This figure demonstrates that granular b-TCP
is obtained, but the arrangement mode is similar to that
of the platelike crystals. This result is due to the granular
b-TCP crystals being converted from the needlelike
ones. Figure 7(h) shows the enlarged view of Fig-
ure 7(g), revealing that the b-TCP crystals are about
1 lm in size. This result corresponds to that for a
calcium phosphate obtained from a calcined sample at a
higher temperature.
[34]
Figure 8 shows the TEM micrographs and ED
patterns of the specimen deposited at 333 K (60 C)
and 10 V under 80 Torr for 1 hour and calcined at
various temperatures for 4 hours. The bright-field (BF)
and ED patterns of the as-deposited sample are shown
in Figures 8(a) and (b). In Figure 8(a), the crystal has a
needlelike shape, 50-nm wide and 150-nm long, with
rough edges. The mechanism of the transformation of
DCPD to HAP can be suggested to occur at the DCPD/
solution interface, where continuous dissolution of
METALLURGICAL AND MATERIALS TRANSACTIONS A VOLUME 41A, DECEMBER 2010—3513
DCPD occurs. Through this dissolution, the interface
experiences an enrichm ent of Ca
2+
and PO
4
3–
ions,
when thermodynamic equilibrium is reached and HAP
is precipitated at the precursor DCPD surface.
[35]
As a
consequence of this reaction, the final microstructure is
constituted of nanosized HAP crystallites. Morpholog-
ical analysis shown in Figure 8(a) suggests that the
DCPD crystals serve as a substrate for the oriented
reprecipitation of the HAP crystallites. Figure 8(b)
shows that the ED ring pattern indexing corresponds
to that of the HAP polycrystals.
The TEM micrograph and ED pattern for a sample
calcined 1073 K (800 C) for 4 hours are shown in
Figures 8(c) and (d), respectively. In Figure 8(c), the
well-formed submicron grains of hexagonal-like b-TCP
are indentified as a wurtzite. The b-TCP was obtained
due to the partial HAP decomposition following Reac-
tion [12] at temperatures above 1073 K (800 C).
[31]
In
addition, b-TCP also formed according to Reaction [15]
at 1063 K (790 C).
[36]
Therefore, 1073 K (800 C) was
chosen as the calcination tempe rature of the deposits to
obtain the b-TCP phase. Ban and Hasegawa
[12]
noted
that the surface process in crystal growth depends on
whether the crystal face is rough or smooth at the
atomic level. Since the HAP transformed to b-TCP at
1073 K (800 C), the surface temperature is higher than
the approximate transition temperature or the driving
force is high. The crystal facet is in roughened state due
to the formation of corners of crystals of b-TCP. The
ED pattern of one submicron grain, as shown in
Fig. 7—SEM micrographs of the deposited samples calcined at various temperatures for 4 h: (a) as-deposited, (b)673K(400C), (c)873K(600C),
(d) enlarged view of (c) indicated by the white circle, (e) 1073 K (800 C), (f) enlarged view of (e) indicated by the white circle, (g) 1273 K (1000 C),
and (h) enlarged view of (g) indicated by the white circle.
3514—VOLUME 41A, DECEMBER 2010 METALLURGICAL AND MATERIALS TRANSACTIONS A

Figure 7(d), was indexed as zone axis = [110] of the
b-TCP.
Figures 8(d) through (f) show the BF image of the
fibrillar grains and ED pattern, respectively. In
Figure 8(e), the onset of crystallization with the forma-
tion of fiberlike CPP crystals is clearly visible. The
dimensions of the fiberlike CPP crystals demonstrate
the decomposition from flap and are 80-nm wide and
400-nm long. Figure 8(f) shows that the ED pattern also
provides the criteria for the presence of CPP in a calcium
phosphate deposited on Ti-6H-4V substrate and after
being calcined at 1273 K (1000 C) for 4 hours. In
general, the fibrous morphology represents a closer
approach to an equilibrium structure, which has trans-
formed slowly. Moreover, the CPP is obtained when the
deposits are calcined at 1273 K (1000 C) for 4 hours
and when there is a high driving force. In contrast,
the interphase precipitation and disl ocation nucleated
Fig. 8—TEM micrograph and ED patterns of the specimen-deposited samples calcined at various temperatures for 4 h: (a) BF image of
as-deposited sample, (b) ED pattern indexing corresponding to HAP, (c) BF image of deposited sample calcined at 1073 K (800 C) for 4 h,
(d) ED pattern corresponding to the b-TCP with ZA = [110], (e) BF image of deposited sample calcined at 1273 K (1000 C) for 4 h, and
(f) ED pattern indexing corresponding to CPP.
METALLURGICAL AND MATERIALS TRANSACTIONS A VOLUME 41A, DECEMBER 2010—3515
structures occur more readily in rapidly transforming
ceramic materials.
IV. CONCLUSIONS
Calcium phosphate was deposited in a 0.04 M
Ca(H
2
PO
4
)
2
ÆH
2
O (MCPM) solution on a Ti-6Al-4V
substrate at 333 K (60 C), 10 V, and 80 Torr for 1 hour
and calcined at various temperatures for 4 hours. The
effect of heat treatment on the calcium phosphate
deposits was investigated using DTA/TGA, FT-IR,
XRD, SEM, TEM, and ED. DTA results show that
an exothermic reaction peak at 793 K (520 C) can be
attributed to the crystallization of HAP. The weak
broad arc-form continuum of the exothermic reaction
that exists between 1023 K and 1123 K (750 Cand
850 C) is due to the formation of calcium pyrophos-
phate (Ca
2
P
2
O
7
, CPP) and b-tricalcium phosphate
[Ca(PO
4
)
2
, b-TCP]. The XRD results show that the
as-deposited sample contains phases of DCPD and
HAP. When a sample is calcined at 1073 K (800 C) for
4 hours, the crystallized phases are composed of the
major phases of b-TCP and CPP, and minor phases of
HAP, CaO, anatase, and rutile. When the deposited
sample is calcined at 1273 K (1000 C) for 4 hours, the
reflection intensity of HAP and CPP decreases, but that
of b-TCP increases. The surface image of the
as-deposited sample shows that the DCPD crystals have
a platelike morphology with a smooth, flat, and sharp
edge. After being calcined at 873 K (600 C) for
4 hours, the morphology of HAP crystals becomes
platelike. Granular b-TCP is also observed, which is
caused by the granular b-TCP crystals being converted
from the platelike crystals.
ACKNOWLEDGMENTS
The authors acknowledge the financial support pro-
vided by the National Science Council Taiwan,
Republic of China (Contact No. NSC93-2216-E-151-
005). We also thank Mr. H.Y. Yao for TEM/EDS
experiments, Mr. F.C. Wu for SEM photography, and
Professor M.P. Hung for suggestions on the manu-
script preparation.
REFERENCES
1. R.J. Friedman, T.W. Bauer, K. Grag, M. Jiang, Y.H. An, and
R.A. Draughn: J. Appl. Biomater., 1993, vol. 14, pp. 661–66.
2. H. Oonishi: Biomaterials, 1991, vol. 12, pp. 171–78.
3. H. Oonishi, S. Kushitani, E. Yasukawa, H. Iwaka, L.L. Hench,
J. Wilson, E. Tsuji, and T. Sugihara: Clin. Orthop., 1997, vol. 334,
pp. 316–25.
4. C.E. Misch and F. Dietsh: Implant Dentistry, 1993, vol. 2,
pp. 158–67.
5. B. Koch, J.G.C. Wolke, and K. de Groot: J. Biomed. Mater. Res.,
1990, vol. 24, pp. 665–67.
6. L.G. Ellies, D.G. Nelson, and J.D. Featherstone: Biomaterials,
1992, vol. 13, pp. 313–16.
7. P. Ducheyne, S. Radin, M. Heughebaert, and J.C. Heughebaert:
Biomaterials, 1990, vol. 11, pp. 244–54.
8. I. Zhitomirsky: Mater. Lett., 2000, vol. 42, pp. 262–71.
9. T.V. Vijayaraghavan and A. Bensalem: J. Mater. Sci. Lett., 1994,
vol. 10, pp. 1782–85.
10. M.H.P. Da Silva, J.H.C. Lima, G.A. Soares, C.N. Elias, M.C. de
Andrade, S.M. Best, and I.R. Gibson: Surf. Coat. Technol., 2001,
vol. 137, pp. 270–76.
11. S.H. Wang, W.J. Shih, W.L. Li, M.H. Hon, and M.C. Wang:
J. Euro. Ceram. Soc., 2005, vol. 25, pp. 3287–92.
12. S. Ban and J. Hasegawa: Biomaterials, 2002, vol. 23,
pp. 2965–72.
13. M. Mansol, G. Jime
´
nez, C. Morant, P. Herrero, and J.M.
Maetı
´
nez-Duart: Biomaterials, 2000, vol. 21, pp. 1755–61.
14. J.S. Chen, H.Y. Jaung, and M.H. Hon: J. Mater. Sci.: Mater.
Med., 1998, vol. 9, pp. 297–300.
15. G. Daculsi: Biomaterials, 1998, vol. 19, pp. 1473–78.
16. J.M. Zhang, C.J. Lin, Z.D. Feng, and Z.W. Tian: J. Electroanal.
Chem., 1998, vol. 452, pp. 235–40.
17. M. Schirkhanzaden: J. Mater. Sci. Lett., 1991, vol. 10, pp. 1415–
17.
18. M. Schirkhanzaden: J. Mater. Sci.: Mater. Med., 1998, vol. 9,
pp. 67–72.
19. V.S. Joshi and M.J. Joshi: Cryst. Res. Technol., 2003, vol. 39,
pp. 817–21.
20. M. Manso, M. Langlet, C. Jime
´
nez, and J.M. Martı
´
nez-Duart: Int.
J. Inorg. Mater., 2001, vol. 3, pp. 1153–55.
21. C. Morterra, A. Chiorino, and A. Zecchina: Gazz. Chim. Ital.,
1979, vol. 109, pp. 683–90.
22. C. Combes, C. Rey, and M. Freche: Coll. Surf. B: Biointerfaces,
1998, vol. 11, pp. 15–27.
23. A. Stoch, A. Brozek, S. Blazewicz, W. Jastrzebski, J. Stoch, A.
Adamczky, and I. Ro
´
j: J. Molec. Struct., 2003, vols. 651–653,
pp. 389–96.
24. K.L. Elmore and T.D. Farr:
Ind. Eng. Chem., 1940, vol. 32,
pp. 580–86.
25. M.J. Sienko and R.A. Plane: in Chemistry: Principles and Prop-
erties, McGraw-Hill, Inc., New York, NY, 1996, p. 512.
26. N. Eliaz and M. Eliyaahu: J. Bio. Mater. Res. A, 2006, vol. 80,
pp. 621–34.
27. K. de Groot: Bioceramics of Calcium Phosphate, CRC Press Inc.,
Boca Raton, FL, 1983, pp. 1–32, 79–97.
28. P.N. De Aza, F. Guitian, A. Merlos, E. Lora-Tamayo, and S. De
Aza: J. Mater. Sci.: Mater. Med., 1996, vol. 7, pp. 399–402.
29. F.C.M. Driessens: in Bioceramics of Calcium Phosphate,K.de
Groot, ed., CRC Press Inc., Boca Raton, FL, 1983, p. 2.
30. J. Zhou, X. Zhang, J. Chen, S. Zeng, and K. de Groot: J. Mater.
Sci.: Mater. Med., 1993, vol. 4, pp. 83–85.
31. K. Kamiya, T. Yoko, K. Tanaka, and Y. Fujiyama: J. Mater. Sci.,
1989, vol. 24, pp. 827–32.
32. L.M. Rodrı
´
guez-Lorenzo, M. Vallet-Regı
´
, and J.M.F. Ferreira:
Biomaterials, 2001, vol. 22, pp. 585–88.
33. F.H. Lin, C.C. Lin, C.M. Lu, H.C. Liu, J.S. Sun, and C.Y. Wang:
Biomaterials, 1995, vol. 16, pp. 793–802.
34. N. Koc, M. Timucin, and F. Korkusuz: Ceram. Int., 2004, vol. 30,
pp. 205–11.
35. M.H. Prado Da Silva, J.H.C. Lima, G.A. Soares, C.N. Elias, M.C.
de Andrade, S.M. Best, and I.R. Gibson: Surf. Coat. Technol.,
2001, vol. 137, pp. 270–76.
36. I.R. Gibson, I. Rehman, M.S. Best, and W. Bonfield: J. Mater.
Sci.: Mater. Med., 2000, vol. 12, pp. 799–804.
3516—VOLUME 41A, DECEMBER 2010 METALLURGICAL AND MATERIALS TRANSACTIONS A
